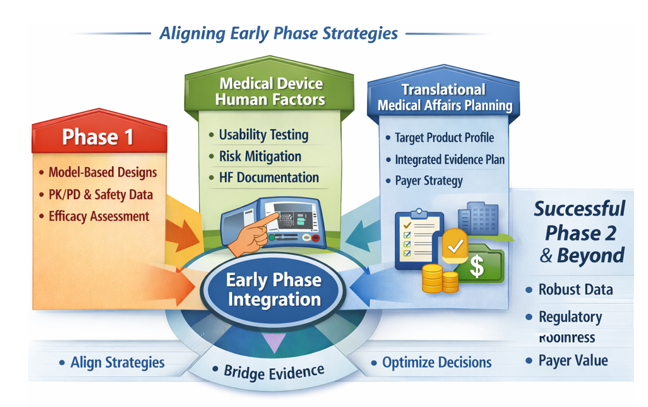
This practical pointer synthesizes three upstream development priorities: Oncology, rare disease, and complex immunology dose‑finding studies, Medical‑device human factors, and Translational Medical Affairs (TMA) evidence planning. This is incorporated into a single, actionable framework for sponsors preparing early‑phase protocols and regulatory submissions and want to get an early read on efficacy.
Phase 1 Oncology (as well as Rare Disease and Autoimmune) Clinical Trials: The 3+3 Default is no Longer Enough for Selecting a Dose
Modern early‑phase oncology development has shifted. The FDA’s January 2026 draft Bayesian guidance explicitly addresses oncology dose‑finding and notes that targeted therapies often require optimized dose selection, not simply identification of the maximum tolerated dose (MTD).[1] As a result, Bayesian and model‑assisted designs: CRM, BLRM, BOIN, mTPI/mTPI2, and efficacy‑toxicity designs are no longer academic alternatives; they are increasingly expected in early oncology programs.[2]
· CRM: Continual Reassessment Method: a model‑based design that continuously updates toxicity estimates to guide escalation more efficiently than rule‑based methods.
· BLRM: Bayesian Logistic Regression Model: a Bayesian dose‑toxicity model (often with escalation with overdose control (EWOC)) that provides flexible, probabilistic control of escalation decisions.
· BOIN: Bayesian Optimal Interval design: a model‑assisted design using optimized decision boundaries for escalate/stay/de‑escalate choices; simpler than CRM/BLRM but statistically stronger than 3+3.
· mTPI / mTPI‑2: Modified Toxicity Probability Interval: Bayesian interval‑based designs that classify doses as below, near, or above the target toxicity rate; mTPI‑2 improves decision consistency.
· Efficacy–toxicity designs: designs that jointly model both efficacy and toxicity to identify an optimal biological dose (OBD), not just the maximum tolerated dose (MTD).
A traditional 3+3 design begins by treating three patients at the starting dose and observing whether any dose‑limiting toxicities (DLTs) occur. Based on the number of DLTs, the next step is predetermined: escalate, expand to six patients, or stop and declare the previous dose the maximum tolerated dose. While familiar, this design provides limited information for modern agents and does not inseparably support dose optimization.
Early‑phase programs should justify dose‑escalation design rather than employing the traditional 3+3 model due to its perceived universal acceptance.
The FDAs new 2026 Bayesian draft guidance emphasizes:
Use of model‑assisted or model‑based designs
Inclusion of operating‑characteristic simulations
Clear articulation of how pharmacokinetic/pharmacodynamic (PK/PD), safety, tolerability, and early activity data will be integrated to guide dose selection
Consideration of more than one biologically active dose for optimization rather than defaulting to the highest tolerated dose
Critical Design Questions Before Protocol Finalization
Is the dose-escalation method justified, not simply inherited?
Do simulations demonstrate escalation/de-escalation behavior, target toxicity accuracy, and the probability of selecting an appropriate dose?
How will PK/PD, safety, tolerability, and early activity data be used to guide dose selection?
Is there a plan to evaluate more than one biologically active dose?
Practical Pointer: A traditional 3+3 design can still be proposed, but it should not appear to be the default because it is familiar. Sponsors should document why the selected design is fit for purpose, how it protects subjects, and how it will support Project Optimus; style dose optimization, which requires early, data‑driven evaluation of multiple active doses using PK/PD, exposure/ response, safety, and early activity data before registration trials.[3]
Medical Devices: Human Factors is Now an Upstream Regulatory Decision
The practical shift: Human factors (HF) refers to the systematic evaluation of how real users interact with a device, focusing on use‑related risks, critical tasks, and the potential for use errors that could lead to harm.
FDA’s final human factors guidance for medical device marketing submissions was issued in late May 2026 and establishes a risk-based framework for what human factors information belongs in 510(k), De Novo, premarket approval (PMA), and humanitarian device exemption (HDE) submissions.[1] The core decision is the HF Submission Category:
· Category 1 generally needs a conclusion and high-level summary
· Category 2 needs a rationale that no critical tasks or impacted critical tasks require validation data
· Category 3 needs a full Human Factors Engineering (HFE)/Usability Engineering (UE) report addressing validation testing for critical tasks
FDA also describes a 60-day operationalization window for submissions received before August 1, 2026.[5]
HF1: high-level summary
HF2: Rationale-based report
HF3: Full validation report
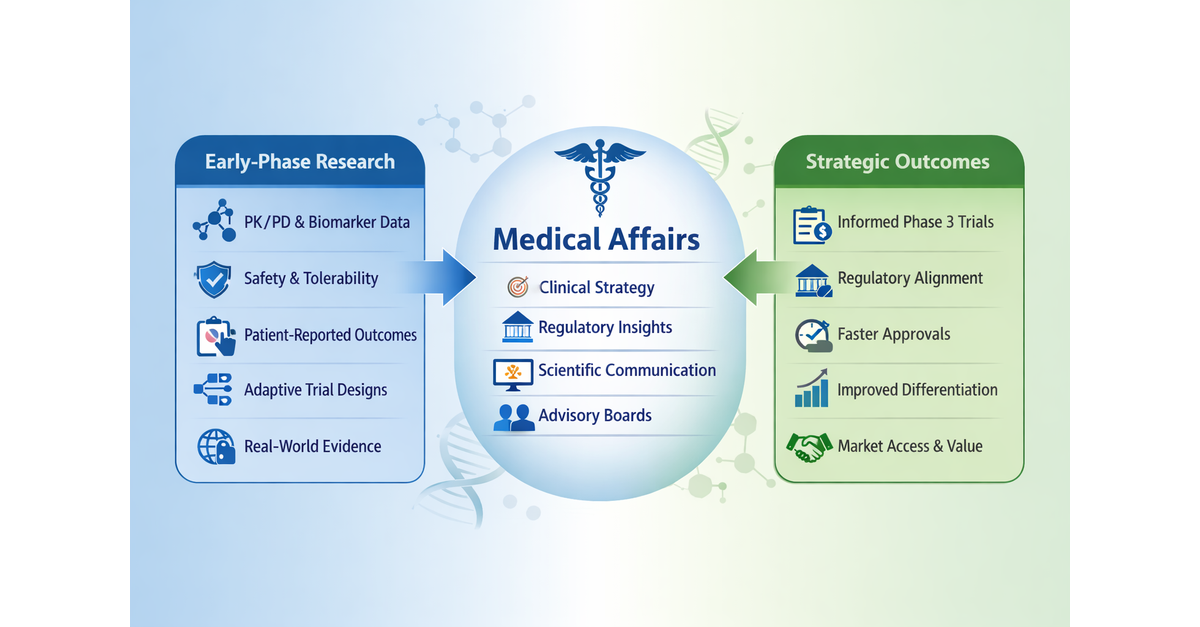
Translational Medical Affairs (TMA): Build the Integrated Evidence Plan Before Phase 2
The practical shift: The Integrated Evidence Plan should be treated as a living development-stage evidence roadmap, initiated no later than end-of-Phase 1/pre-Phase 2 and updated as clinical, regulatory, payer, and external stakeholder assumptions evolve. The IEP should map each decision-maker question (regulator, payer, prescriber, patient, and investigator) to a specific evidence source, endpoint, comparator, method, and timepoint.[6],[7]
Three linked planning documents that teams should evidence generation across development:[8] The TPP defines the intended product profile and claims; the CDP defines the clinical studies needed to support approval; the IEP defines the broader evidence needed to support regulatory, payer, clinician, patient, and post-approval decision-making.
The Target Product Profile (TPP) is a strategic planning tool that defines the intended efficacy, safety, and usability goals for the therapeutic under development. It outlines the key objectives the product must meet at each stage, supports go/no‑go decisions, and anchors development plans to the team’s discussions with regulatory agencies regarding evidence needs for submission.
The Clinical Development Plan (CDP) A plan which includes the overall development strategy, proposed phase 1, phase 2, and phase 3 study designs, dose-selection approach, endpoints, safety monitoring plan, regulatory strategy, statistical considerations, and major decision points. It also aligns clinical activities with nonclinical, CMC, pharmacovigilance, medical affairs, and commercial objectives.
The Integrated Evidence Plan (IEP) a cross‑functional plan that identifies the key questions decision makers will ask and the data needed to answer them. It outlines how clinical, regulatory, commercial, and patient‑ experience evidence will be generated, the timing and methods for producing that evidence within the clinical development plan, and how it will support both the regulatory submission and potential peri and post‑approval questions about real‑world use. The IEP should have explicit governance, with cross-functional input from Clinical Development, Regulatory, Medical Affairs, HEOR, Market Access, Patient Engagement, Biostatistics, Safety, and Medical Communications/Publications, and should be revisited at major data readouts, when new external data (e.g., competitor, regulatory, market) become available, and protocol decision points.
Use phase 2 to pre-position payer evidence
· Employ Disease‑specific patient-reported outcomes (PROs) and health‑utility measures: Use validated patient‑reported outcome tools tailored to the condition being studied, (as much as possible) along with standardized health‑utility instruments such as the SF-36 or EQ‑5D‑5L when relevant. These measures capture how patients feel and function in daily life, quantify symptoms and treatment burden directly from the patient perspective, and provide utility values needed for health‑economic analyses and value assessments.
· Collect healthcare-resource utilization and patient-burden data prospectively.
· Avoid relying solely on post-hoc indirect comparisons when feasible comparators could have been embedded earlier.[9]
Make field medical insights an evidence-planning asset
· Use structured insight capture tied to IEP evidence gaps, not free-text anecdote collection.
· Run a quarterly insights-to-evidence review with documented actions.
· Measure actioned insights, investigator-initiated study (IIS) opportunities, real-world evidence (RWE) analyses, and medical communications/publications, not just interaction counts.
Unified Practical Pointers for Sponsors
Before finalizing the next protocol or submission, sponsors should:
· Justify dose finding design with simulations and dose optimization logic
· Assign the HF submission category as part of the design controls and align with Quality Management system Regulation (QMSR)
· The output of the IEP should not be a static slide deck. It should be a decision tool that identifies which evidence gaps must be closed before phase 2, before pivotal trial initiation, before submission, and before launch.
Treat these as integrated development decisions, not isolated regulatory tasks. The strongest programs will align dose strategy, human-factors planning, and evidence generation early, reducing downstream risk and strengthening regulatory and payer readiness.
References:
[1] U.S. Food and Drug Administration. Use of Bayesian Methodology in Clinical Trials of Drugs and Biologics: Draft Guidance for Industry. Published January 12, 2026.
[2] Yuan J. A practical summary of FDA draft guidance: Use of Bayesian methodology in clinical trials of drug and biological products (January 2026). Published January 18, 2026.
[3] U.S. Food and Drug Administration. Project Optimus. FDA Oncology Center of Excellence. Accessed June 2026. https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus
[4] U.S. Food and Drug Administration. Content of Human Factors Information in Medical Device Marketing Submissions: Guidance for Industry and FDA Staff. Final Guidance. Published May 2026.
[5] U.S. Food and Drug Administration. Final guidance for human factors information in medical device marketing submissions. FDA Bulletin. Published May 28, 2026.
[6] Evidinno Outcomes Research. Integrated Evidence Generation Plans in Modern Biopharma. Published 2025.
[7] Lumanity. Integrated Evidence Generation Planning: From Strategy to Execution. Published 2026.
[8] Medical Affairs Professional Society (MAPS). Strategic Integrated Evidence Generation Planning: Company and Non‑Company Sponsored Research Guidance Document. Published 2024.
[9] Prescient Healthcare Group. Developing a Best‑in‑Class Integrated Evidence Plan: From Strategy to Studies. Published 2024.