Practical Pointers for Drug Development / December 2025
Authors: Gerald L. Klein, MD1; Roger E. Morgan, MD1; Melissa Palmer, MD2; Freddy Byrth1; Michael Fath, PhD3; Katie-Louise Dawson, MD1; Colin Scott, MD4; Patrick Loebs1; Rob Walsh, MD1
Affiliations: MedSurgPI; Liver Consulting, LLC2; Cavabio Consulting3; Highland Airs Consulting4
Drug Development
Early-phase studies now play a larger role for generating clinical, mechanistic, and real-world insights that historically emerged only during phase 3 development.[1] Traditionally, phase 1 studies focused primarily on safety, tolerability, and pharmacokinetics, often conducted in healthy volunteers when appropriate. By incorporating elements relevant to future marketing applications, such as those required for New Drug Application (NDA), Biologics License Application (BLA), or Marketing Authorization Application (MAA) submissions, into phase 1 and phase 2 studies, development teams can generate critical evidence earlier in the program. [1,2] These elements may include response relationships, expanded safety datasets, patient-experience measures, and real-world clinical context. Early integration of these components helps prevent late-stage trials from becoming burdened with variables introduced too late in development, while strengthening alignment with the Target Product Profile (TPP) and the overall Clinical Drug Development Plan. [3]
Phase 1 trials should therefore be designed to provide maximum strategic value, reduce phase 2 failure risk, and strengthen investor and regulatory confidence [1,2]
o Although primarily intended for first-in-human safety and tolerability assessments, phase 1 trials should also function as value-creation platforms that:
§ Incorporate pharmacokinetics (PK) and pharmacodynamics (PD) modeling, when financially and otherwise feasible, to inform dose selection and variability assessment [3]
§ Identify exposure–response relationships early in order to inform phase 2 design[3]
§ Explore multiple dose levels with sufficient sampling density to characterize pharmacologic activity and, for targeted therapies such as oncology or immunomodulatory agents, define the biologically effective or optimal dose.
· Incorporating adaptive or model-informed approaches (including machine learning methods) to accelerate dose optimization and minimize exposure to subtherapeutic dosing. Examples include: [3,4]
o Bayesian dose-escalation designs [4]
o Model-informed drug development (MIDD) strategies aligned with FDA guidance [3]
o Real-time PK analysis for data-driven decision making [3]
o Adaptive cohort expansion to refine dose and population selection4
Importantly, the traditional use of healthy volunteers in phase 1 studies remains appropriate for many therapeutic classes, particularly when the anticipated safety profile permits such evaluation. Healthy volunteer studies allow clearer pharmacokinetic characterization, minimize confounding effects from underlying disease, and provide efficient assessment of tolerability and dose-related safety. This approach remains common for agents such as antihypertensives, antibiotics, metabolic drugs, and many neurological therapies.
However, in some therapeutic areas (oncology, gene therapies, immunotherapies, and other targeted or higher-risk modalities) phase 1 studies are frequently conducted directly in patients because of safety considerations, disease specificity, or the need to observe early signals of biological activity.
Hybrid early-phase designs are increasingly used, in which initial cohorts of healthy volunteers are evaluated for safety and pharmacokinetics, followed by patient cohorts that explore dose escalation, pharmacodynamic effects, and early signals of activity.
· In oncology, rare disease, or high-unmet-need indications, early efficacy-generating components may also be strategically integrated to support expedited development pathways, including: [5,6]
o Expansion cohorts within phase 1 protocols
o Early proof-of-concept arms
o Enrichment strategies (e.g., biomarker-selected populations) to increase the probability of demonstrating a meaningful signal
o Independent medical monitoring should be incorporated into all clinical studies involving human participants so that adequate safety is provided for the trial
· For immunomodulators, gene therapies, biologics, and other first-in-class agents, embed robust safety governance mechanisms consistent with ICH E6(R3) Quality-by-Design principles.
· These mechanisms may include:
o Independent Adjudicators
o Independent Safety Review Committee (SRC)
o Predefined halting/stopping rules, both for individual patients and the clinical trial
o Sentinel dosing for initial human exposure [7]
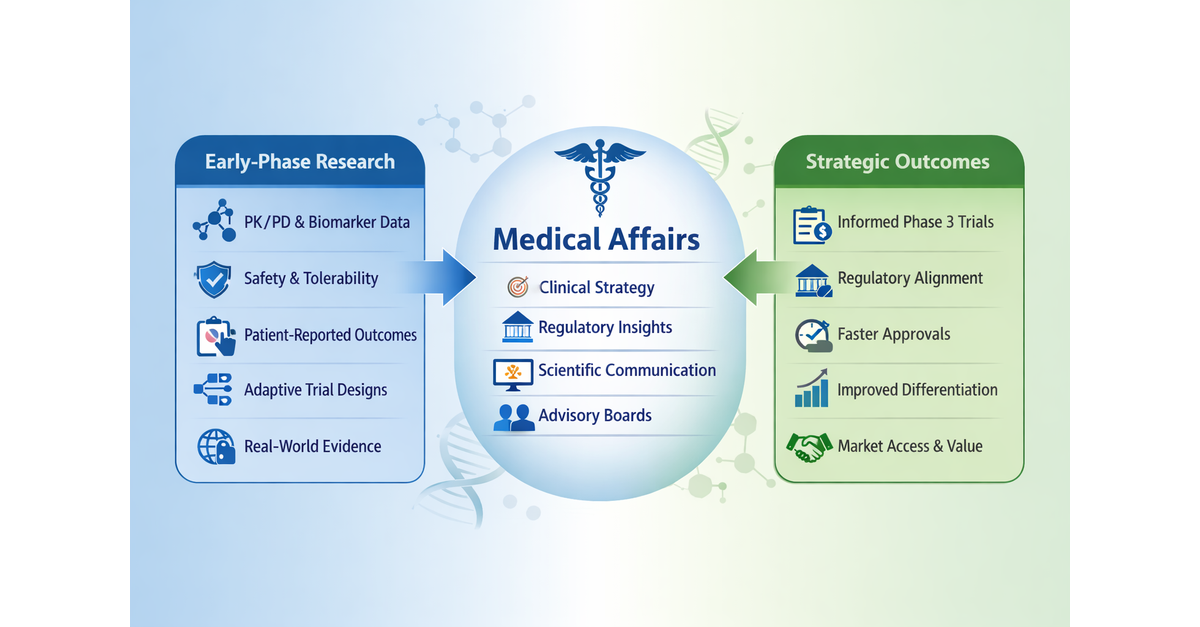
Medical Affairs
Medical Affairs should play a key role in early drug development by helping translate emerging clinical data into scientific, regulatory, and strategic insights. Early engagement supports cross-functional collaboration and the development of a robust TPP that guides clinical design, regulatory planning, and future product positioning. [8]
· Early engagement of Medical Affairs, alongside Regulatory Affairs, helps ensure that emerging clinical data are interpreted and positioned within the broader scientific and clinical context. [9-11] Areas where cross-functional planning is particularly important include:
o Evaluating special-population considerations (renal or hepatic impairment studies) [12]
o Assessing drug-drug interaction potential and food-effect impact proactively [12]
o Conducting QT assessment when relevant (ICH E14 alignment) [13]
o Evaluating immunogenicity risk for biologics and complex modalities [14]
· Medical Affairs also contributes to scientific and clinical advisory board strategy and asset differentiation by engaging internal and external subject matter experts (SMEs) [8-10] including:
o Identifying and engaging appropriate scientific advisory boards
o Helping develop key differentiators such as:
§ Positioning quality-of-life (QoL) endpoints and patient-reported outcomes (PROs) within the broader clinical and value narrative once study results are available[15]
§ Outcomes demonstrating advantages over current therapies
§ Regulatory credibility through alignment with FDA and global agency expectations[16]
§ Risk-reduction strategies that enhance program resilience
§ Strategies that increase asset valuation and partnership appeal
· Development teams should ensure early cross-functional collaboration and accountability to support the creation and refinement of a rigorous TPP.11 Key elements to consider include:
o Incorporating real-world evidence (e.g., observational studies, registries, or healthcare data sources) and early clinical insights from clinical practice and investigators
o Defining the proposed indication
o Specifying the dose and dosing regimen
o Clarifying the route of administration
o Defining the target population
o Anticipating the safety profile
o Articulating differentiation vs. standard of care
o Defining differentiation from competitive products
[1] Morgan P, et al. Impact of a five-dimensional framework on R&D productivity. Nat Rev Drug Discov.2018;17(3):167-181.
[2] Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61(3):275-291.
[3] US Food and Drug Administration. Target Product Profile—A Strategic Development Process Tool. 2007.
[4] Neuenschwander B, Branson M, Gsponer T. Bayesian adaptive dose-escalation designs. Stat Med. 2008;27(13):2420-2439.
[5] Beaver JA, Howie LJ, Pelosof L, et al. A 25-year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics. JAMA Oncol. 2018;4(6):849-856.
[6] US Food and Drug Administration. Expedited Programs for Serious Conditions—Drugs and Biologics: Guidance for Industry. FDA; 2014.
[7] US Food and Drug Administration. Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials. FDA; 2007.
[8] US Food and Drug Administration. Target Product Profile—A Strategic Development Process Tool. FDA; 2007.
[9] O’Connor D, Lee S, et al. The evolving role of medical affairs. Ther Innov Regul Sci. 2019;53(4):447-455.
[10] Engelberg AB, Kesselheim AS. Medical affairs and the shifting pharmaceutical landscape. N Engl J Med. 2016;374:1787-1789.
[11] Garattini S, Padula A, et al. The role of medical affairs in evidence generation. Lancet. 2020;395:1968-1970.
[12] US Food and Drug Administration. Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications: Guidance for Industry. FDA; 2020.
[13] International Council for Harmonisation. E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. 2005. Updated Q&A 2017.
[14] US Food and Drug Administration. Clinical Pharmacology Considerations for Biologics: Guidance for Industry. FDA; 2019.
[15] Basch E, Bennett AV. Patient-reported outcomes in drug development. JAMA. 2017;318(2):197-198.
[16] International Council for Harmonisation. ICH Harmonised Guideline E6(R3): Good Clinical Practice. 2023.