MedSurgPI, LLC is proud to share our editorial: More Physicians and Dentists Should Participate in Clinical Trials Now! This article has been accepted by Cureus. This piece highlights how research involvement can expand patient options, strengthen clinical relevance, and connect everyday practice to emerging science. A sincere thanks to our outstanding coauthors for their expertise, collaboration, and commitment to advancing clinical research across diverse practice settings.

Biobanking: The Hidden Infrastructure Behind Precision Medicine
The East Carolina University (ECU) School of Dental Medicine is building its research enterprise.
This spotlight features a saliva Biobank at the ECU School of Dental Medicine as it builds its research enterprise under Dr. Alexandre R. Vieira, DDS, MS, PhD, Dean for Research
Authors: Gerald L. Klein, MD[1]; Freddy Byrth, BS[1]; Michael Fath, PhD[2]; Aida Carfagno, BS[3]; David Weinstein, MD, PhD[4]; Patrick Loebs, MSW, MSH[1]
Affiliation: MedSurgPI[1]; Cavabio Consulting[2]; Cedar and Stone Consulting[3]; David Weinstein Consulting[4]
Technology Innovation Spotlight features a saliva Biobank at the ECU School of Dental Medicine. Behind nearly every modern biomarker, companion diagnostic, and target-validation program sits an asset that seldom appears in the press release: well-characterized biospecimens, collected under controlled conditions and linked to longitudinal clinical data. Biobanking, the systematic collection, processing, storage, annotation, and distribution of biological specimens and their associated data, has quietly become one of the most consequential enabling technologies in drug, biologic, device, and diagnostic development. What was once thought of as “a freezer in the basement” is now a data-rich, standards-governed, increasingly automated infrastructure layer for precision medicine. Modern biobanks are therefore not only technical infrastructure; they are also trust infrastructure, dependent on transparent consent, privacy protections, and participant confidence that specimens and data will be used responsibly.
THE OPPORTUNITY
Saliva is among the most practical biofluids for population-scale collection. It can be gathered noninvasively, at low cost, and without specialized phlebotomy, and it carries a rich repertoire of analytes, including cells, nucleic acids, extracellular vesicles, metabolites, and proteins. Because oral and systemic health are increasingly understood to be connected, saliva offers a window not only into caries and periodontal disease but also into systemic conditions, with the added advantage of supporting repeated, longitudinal sampling at minimal patient burden.
THE INNOVATION
The center of gravity in biobanking has shifted from the specimen to the specimen plus the data wrapped around it. Three converging developments explain the change.
First, scale. Population-scale biobanks now operate at a size that was impractical a decade ago. The UK Biobank enrolled roughly 500,000 participants with linked genomic, imaging, and health-record data,[1] while the U.S. All of Us Research Program was designed explicitly to build a large, diverse cohort that mirrors the populations medicine actually serves.[2]
Second, data linkage. The research value of a biobank today depends heavily on how richly its specimens connect to electronic health records that can be queried in depth; robust phenotyping, and multi-omic readouts. The combination, not the specimen in isolation, is what enables agnostic and unbiased discovery across thousands of associations among diseases, genes, and exposures.[3]
Third, quality as a discipline. Preanalytical variables such as time to processing, freeze and thaw cycles, fixation, and storage temperature can materially alter the molecular characteristics of a sample and, with it, the validity of any downstream result. Reporting frameworks such as BRISQ (Biospecimen Reporting for Improved Study Quality) were created to make those variables transparent and reproducible,[4] and the international standard ISO 20387 now defines general requirements for biobanking competence and consistency.[5] Alongside scale and linkage, this quality science is what turns a collection of tubes into an extensive and rich research asset.

WHY IT MATTERS
• Noninvasive, low-cost collection enables large, diverse cohorts and repeat sampling that blood-based biobanks struggle to match.
• Chart-linked saliva supports biomarker discovery for oral conditions such as caries and periodontitis, as well as for systemic disease through the oral-systemic axis.
• A newer school building its portfolio can design consent scope, quality systems, and annotation for partnering from the outset rather than retrofitting them later.
• For sponsors, a well-governed saliva resource offers a practical platform for noninvasive biomarkers and companion-diagnostic work.
Representative large-scale biobanks frequently cited as exemplars:

FEATURED EXAMPLE:
Saliva is among the most practical biofluids for population-scale collection. It can be gathered noninvasively, at low cost, and without specialized phlebotomy, and it carries a rich repertoire of analytes, including cells, nucleic acids, extracellular vesicles, metabolites, and proteins. Because oral and systemic health are increasingly understood to be connected, saliva offers a window not only into caries and periodontal disease but also into systemic conditions, with the added advantage of supporting repeated, longitudinal sampling at minimal patient burden. ECU School of Dental Medicine is building its research enterprise under Dr. Alexandre R. Vieira, DDS, MS, PhD, Dean for Research, a dentist and human molecular geneticist whose work emphasizes characterizing patients’ whole health trajectories rather than studying one disease at a time. At his prior institution, Dr. Vieira established a dental-school registry under which patients entering the building were invited to contribute two things: permission to draw data from their records and a saliva sample. That pairing of biological specimen with chart-linked clinical data is the linkage that gives a modern biobank its translational power, and it is the model now being extended at ECU as the school formalizes a research portfolio supported by its Office of research facilities, including a Basic Science Laboratory, a Vivarium, and a Clinical Research Center.[1]
Why this matters to sponsors and licensees:
· Specimen quality is a data-quality problem: undocumented preanalytical variables undermine biomarker and companion-diagnostic claims.
· Consent scope is a gating factor: commercial and future-use permissions determine whether a collection can support a development program.
· ISO 20387 accreditation and BRISQ-style reporting are now table stakes in diligence, not nice-to-haves.
Diverse, electronic health record-linked cohorts shorten the path from hypothesis to validated, generalizable evidence.
THE COMMERCIAL AND TRANSLATIONAL OPPORTUNITY
Industry licensees and development partners do not value specimens generically. They value fit-for-purpose specimens. Four attributes drive whether a collection is commercially useful: documented provenance and preanalytical history; the depth and accuracy of clinical annotation; the scope of consent (in particular, whether commercial and future unspecified research use is permitted); and quality accreditation against an industry-recognized standard. Fit for purpose also depends on intended use: a collection that is adequate for exploratory biomarker discovery may not be sufficient for clinical validation, companion diagnostic development, or regulatory submission if chain of custody, preanalytical controls, assay validation, consent scope, or population representativeness are incomplete. A technically impressive collection with ambiguous consent or undocumented handling can be commercially unusable, an avoidable and expensive surprise late in a program.
For institutions that hold biobank assets, including universities, academic medical centers, and federal labs, this creates a real partnering opportunity, provided the asset is positioned in the language sponsors evaluate against. That framing is where many otherwise-strong collections under-realize their value.
REGULATORY, ETHICAL, AND QUALITY CONSIDERATIONS
Biobanking sits at the intersection of human-subjects protection, privacy law, and quality systems. In the United States, the revised Common Rule introduced provisions for broad consent covering storage, maintenance, and secondary research use of identifiable specimens and data,[1] while HIPAA governs the handling of protected health information. In the EU, GDPR imposes its own constraints on personal-data processing. Specimens used to support regulated product validation, particularly diagnostic and companion-diagnostic validation, additionally invite FDA scrutiny of provenance, characterization, and fitness for the intended use. Layered on top are quality and competence standards (ISO 20387) and BRISQ recognized biorepository best practices. The practical takeaway: governance and documentation decisions made at the moment of collection determine, years later, whether a specimen can be used at all.
MedSurgPI works at the intersection of clinical development, regulatory strategy, and Translational Medical Affairs, the same intersection where biobank value is won or lost. For institutions and inventors, we translate the science of a collection into the terms industry licensees actually evaluate: provenance, consent scope, fitness for purpose, and the regulatory path the specimens can support. For sponsors, we help define fit-for-purpose specimen and annotation requirements up front, scope consent to the development objective, and avoid the late-stage discovery that a collection cannot support the claim it was meant to enable. Medical Affairs adds particular value by defining the clinically meaningful questions a biobank should answer, identifying KOL and sponsor use cases, shaping evidence‑generation plans, and communicating both the potential and the limitations of the asset transparently.
Practical Pointer
A biobank’s translational ceiling is set at the moment of consent, collections built with commercial and future-use permissions from day one avoid the costly retrofitting that sidelines otherwise strong assets.

[1] Office for Human Research Protections, US Department of Health and Human Services. Federal Policy for the Protection of Human Subjects; Final Rule. Fed Regist. 2017;82(12):7149-7274. Codified at 45 CFR part 46.
[2] East Carolina University School of Dental Medicine. Research. Accessed June 9, 2026. https://dental.ecu.edu/research-department/
[3] Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. doi:10.1371/journal.pmed.1001779.
[4] All of Us Research Program Investigators. The “All of Us” Research Program. N Engl J Med. 2019;381(7):668-676. doi:10.1056/NEJMsr1809937.
[5] Beesley LJ, Salvatore M, Fritsche LG, Pandit A, Rao A, Brummett C, Willer CJ, Lisabeth LD, Mukherjee B. The emerging landscape of health research based on biobanks linked to electronic health records: existing resources, statistical challenges, and potential opportunities. Stat Med. 2020;39(6):773-800. doi:10.1002/sim.8445.
[6] Moore HM, Kelly AB, Jewell SD, McShane LM, Clark DP, Greenspan R, Hayes DF, Hainaut P, Kim P, Mansfield E, Potapova O, Riegman P, Rubinstein Y, Seijo E, Somiari S, Watson P, Weier HU, Zhu C, Vaught J. Biospecimen reporting for improved study quality (BRISQ). J Proteome Res. 2011;10(8):3429-3438. doi:10.1021/pr200021n.
[7] International Organization for Standardization. ISO 20387:2018. Biotechnology, Biobanking, General Requirements for Biobanking. Geneva, Switzerland: International Organization for Standardization; 2018.
Oral Health: A Missing Piece of Routine Care
From East Carolina School of Dentistry Research Division and MedSurgPI
Authors: Gerald L. Klein, MD1; Alexandre R. Vieira, DDS, MS, PhD2; Stephen Haworth, MB, BS1; Freddy Byrth, BS1; Roger Morgan, MD1; Rob Walsh, MD1; Larry Florin, MBA3
Affiliations: MedSurgPI, LLC1; East Carolina University School of Dental Medicine2; LBF Biopharma Consulting3
Why Primary Care Should Ask, Look, Treat, and Refer
Practical pointer: A one-minute oral-health questionnaire paired with a quick visual check of the mouth helps primary care teams identify pain, infection, cancer warning signs, dry mouth, caries risk, periodontal risk, and lack of dental access. The goal is not to make primary care a dental office; it is to close a common gap by counseling, applying evidence-supported preventive care when appropriate, treating urgent medical consequences, and arranging timely dental referrals.
The mouth is part of the body, but it is often left outside routine medical care. That separation matters. CDC surveillance data show that nearly 21% of U.S. adults aged 20-64 had untreated decay in permanent teeth, with higher burden among people living in poverty, some racial and ethnic groups, (for example, American Indian/Alaska Native, African American and other historically marginalized populations), people with lower educational attainment, and current smokers.[1] Untreated oral disease can produce pain, infection, tooth loss, difficulty eating and speaking, missed school and work, and avoidable emergency visits.[2]
Primary care is uniquely positioned to help. Patients with diabetes, pregnancy, xerostomia, tobacco or alcohol use, immunosuppression, chronic disease, medication burden, or poor access to dental care are already in our exam rooms. A simple oral-health workflow can be framed as Ask, Look, Decide, Act, and Document/Follow Up - a model described for primary care integration by the National Academy of Medicine and the Oral Health Delivery Framework.[3]
This is a practical clinical screen, not a substitute dental diagnosis. The United States Preventive Services Task Force (USPSTF) found insufficient evidence to recommend for or against routine primary-care screening or preventive interventions for asymptomatic adults and for children/adolescents aged 5-17. That insufficient “evidence statement” should not be read as a recommendation to ignore the mouth; it means the evidence is incomplete and clinicians should use judgment. However, Rural America has a greater incidence of oral problems and recommends a greater need for this intervention.[4] The stronger primary care role is to identify symptoms and risk, deliver brief prevention, and connect patients to dental care. For children younger than 5 years, the USPSTF specifically recommends fluoride varnish after primary tooth eruption and oral fluoride supplementation from age 6 months when the water supply is fluoride deficient.[5],[6]

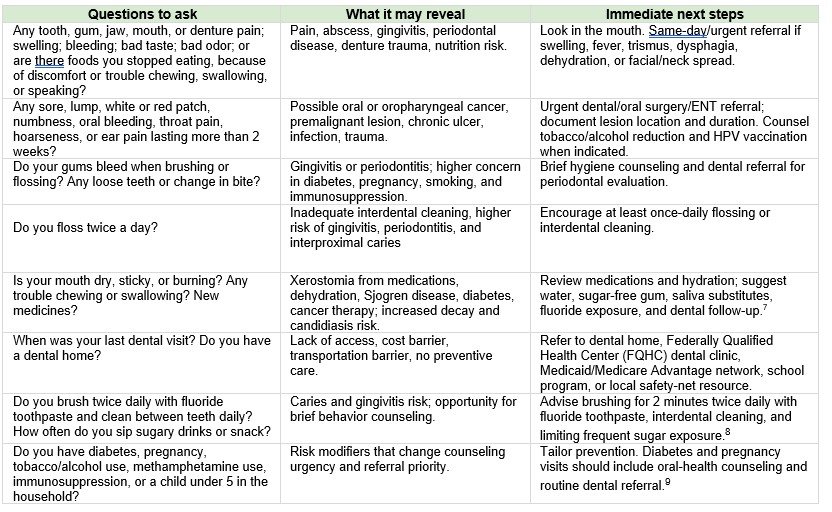
A short questionnaire for primary care
Use these questions at annual visits, well-child visits, diabetes visits, pregnancy visits, medication reviews, transitions of care, and visits for pain, fever, swelling, weight loss, or nutrition problems. A positive answer should prompt a brief mouth exam and a clear follow-up plan.
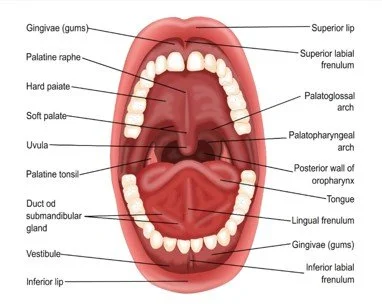
The brief mouth look
Use gloves, a light, and a tongue blade. Inspect the lips, buccal mucosa, tongue and floor of mouth, palate, gingiva, teeth, dentures, and visible oropharynx. Note saliva quality, odor, plaque, visible decay, broken teeth, gingival swelling, ulcers, white patches that scrape or do not scrape, red patches, masses, facial swelling, trismus, and ability to swallow. If clinic policy allows photos, obtain consent and include images in the referral. CDC lists oral-cancer symptoms such as nonhealing sores, swelling or unusual bleeding, difficulty breathing/speaking/ chewing/swallowing, and persistent mouth or throat pain; tobacco and alcohol are common risk factors, and HPV is linked to some oropharyngeal cancers.[1]
Preventive and therapeutic actions
Most oral diseases require definitive dental care. Primary care can still reduce harm by triaging urgency, treating medical complications, and making referral specific rather than vague.
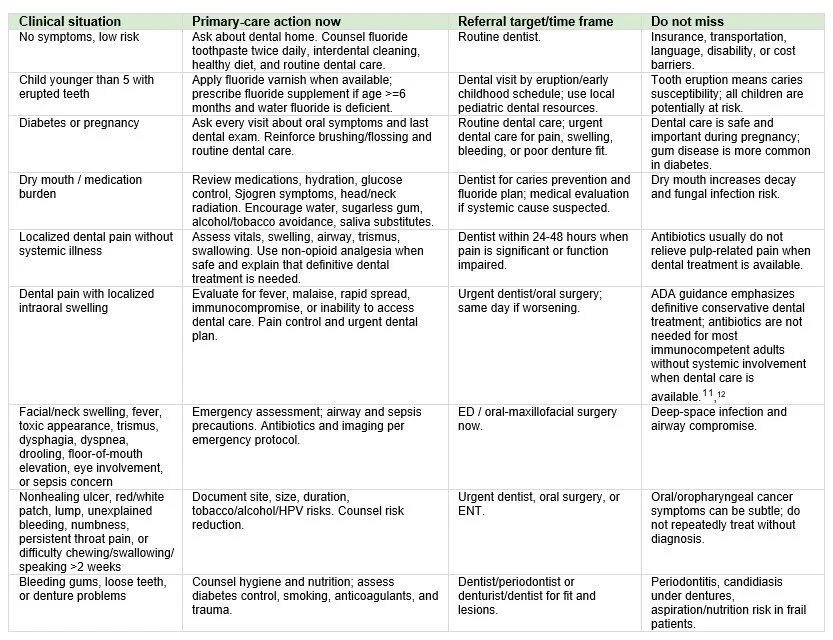
Make referral closed-loop, not aspirational
A strong oral-health plan is specific: name the concern, urgency, destination, and follow-up. For example: “localized dental swelling without fever, urgent dental appointment within 48 hours; call if fever, worsening swelling, difficulty swallowing, or breathing symptoms.” Build a dental referral list that includes practices accepting Medicaid, Federally Qualified Health Center (FQHC) dental sites, pediatric dentists, oral surgeons, ENT clinics, pregnancy-capable dental practices, and after-hours emergency pathways. Document the questionnaire response, mouth findings, treatment/counseling, referral, and how the team will confirm completion. Because oral disease contributes to significant productivity and school losses nationally, prevention and care navigation should be considered essential elements of primary care, not add-ons.[1]
Oral cancer: An estimated 60,500 new cases will be diagnosed in 2026.[2] They are usually diagnosed late; primary care examination may help to modify this current situation.
Bottom line: Primary care should evaluate the mouth because oral problems are common, visible, consequential, and often missed. A short questionnaire, a quick look, brief prevention, targeted therapy, and closed-loop referral can prevent avoidable suffering while respecting the essential role of dental professionals.
[1] Centers for Disease Control and Prevention. Oral Health Facts. May 15, 2024. https://www.cdc.gov/oral-health/data-research/facts-stats/index.html
[2] CDC, 2024 Oral Health Surveillance Report: Selected Findings; USPSTF, Oral Health in Adults: Screening and Preventive Interventions, 2023.
[3] National Academy of Medicine, Integration of Oral Health and Primary Care: Communication, Coordination and Referral; Oral Health Delivery Framework: Ask, Look, Decide, Act, Document & Follow-Up.
[4] U.S. Preventive Services Task Force. USPSTF Procedure Manual. 2021. Accessed May 13, 2026. https://www.uspreventiveservicestaskforce.org
[5] USPSTF, Prevention of Dental Caries in Children Younger Than 5 Years: Screening and Interventions, 2021.
[6] USPSTF, Oral Health in Adults: Screening and Preventive Interventions, 2023; USPSTF, Oral Health in Children and Adolescents Aged 5 to 17 Years, 2023.
[7] National Institute of Dental and Craniofacial Research, Dry Mouth.
[8] American Dental Association, Home Oral Care, updated 2024; American Dental Association, Topical Fluoride Clinical Practice Guideline, 2013.
[9] CDC, Promoting Oral Health for People With Diabetes, 2024; CDC, Dental Care Is Safe and Important During Pregnancy, 2024.
[10] CDC, About Oral Cancer, 2024.
[11] American Dental Association, Antibiotics for Dental Pain and Swelling Guideline, 2019; endorsed by the American College of Emergency Physicians.
[12] American Dental Association. Antibiotics for Dental Pain and Swelling Guideline. 2019. https://www.ada.org/resources/research/science/evidence-based-dental-research/antibiotics-for-dental-pain-and-swelling
[13] CDC, Oral Health Facts, 2024.
[14] American Cancer Society. Key Statistics for Oral Cavity and Oropharyngeal Cancers. Updated March 23, 2026.
Medical Monitoring is Critical: The Industry is Ready for a Standard

LinkedIn poll results suggest interest in a “formal certification process”
Authors: Gerald L. Klein, MD[1]; Roger Morgan, MD[1]; Melissa Palmer, MD[2]; Freddy Byrth, BS[1]; Patrick Loebs, MSW, MPH[1]; Stephen Haworth, MB[1]; Emila Jones, MD[3]; Katie Louise Dawson, MD[1]
Affiliations: MedSurgPI[1]; Liver Consulting, LLC[2]; EJ Med Insights[3]
Medical monitoring operates within one of the most consequential intersections in clinical research: participant safety, protocol integrity, regulatory compliance, clinical judgment, and real-time operational decision-making. Yet despite its importance, the role remains inconsistently defined across sponsors, clinical research Organizations (CROs), therapeutic areas, and study phases and types.
For the purposes of this discussion, medical monitoring is defined as medically qualified oversight that supports participant safety, protocol interpretation, participant eligibility, medical-data review, safety surveillance, escalation of emerging risks, and clinical decision support to sponsors, CROs, investigators, and safety governance bodies.
We recently surveyed the clinical research community in a LinkedIn poll through the following question with one of four possible responses. To ensure the poll reached the appropriate audience, we distributed it specifically within LinkedIn groups composed of clinical research professionals. Although this represents a relatively small sample of the broader clinical trial community, we believe the responses offer a meaningful anecdotal reflection of prevailing community perspectives.

That is a meaningful signal. While the sample size is limited, the results suggest meaningful professional recognition of a growing gap in standardization. The broader conversation is shifting from whether medical monitoring matters to how it should be defined, supported, and documented. It is now a question of whether the industry should define a consistent standard, who performs medical monitoring, what competencies are required, and how readiness is demonstrated.
Why does this question matter now?
Clinical trials are becoming increasingly complex across scientific, operational, and regulatory dimensions. Protocols now frequently incorporate adaptive designs, decentralized elements, biomarker‑defined populations, digital endpoints, real‑world data, sophisticated safety requirements, global execution models, and intricate monitoring or stopping criteria. These factors, combined with substantially higher data volumes, create significant burden for both participants and research sites. Recent analyses from the Tufts Center for the Study of Drug Development (CSDD) and TransCelerate BioPharma both confirm that Phase III pivotal trials now involve far more procedures and data points than in previous decades, underscoring the accelerating complexity of modern clinical research.²
Table 1 summarizes these drivers of complexity and their operational impact on sites and participants.
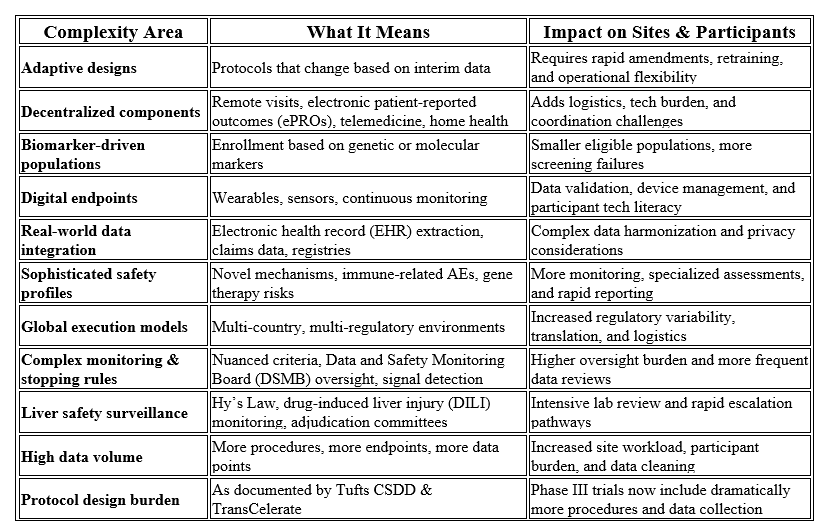
At the same time, global regulatory expectations are moving toward risk-based quality management, proportionality, quality by design, and clearer accountability. The new International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E6(R3) Good Clinical Practice (GCP) guidance emphasizes flexible, risk-based approaches, quality by design, participant protection, reliability of trial results, and clarified sponsor and investigator responsibilities.³ The FDA’s risk-based monitoring guidance similarly focuses sponsor oversight on the most important aspects of study conduct and reporting to enhance human subject protection and data quality.⁴
In other words, the industry is being asked to become more agile and adopt more risk-based operational strategies, while trials are becoming more medically, operationally, and technologically complex. Medical monitoring expertise is central to that transition to more complex studies and greater safety oversight.
The standardization gap
Current regulations and guidance establish sponsor accountability for appropriate monitoring, safety review, and use of qualified personnel. For example, FDA regulations state that sponsors are responsible for ensuring proper monitoring of investigations, and that sponsors must select monitors qualified by training and experience.⁵ The National Institutes of Health (NIH) policy also recognizes the medical monitor as a possible data and safety monitoring entity and states that monitoring plans should be commensurate with the level of risk, nature and complexity of the research, and population under study.⁶
However, regulators generally do not define a standardized role-specific set of competencies for the modern medical monitor, and current expectations do not translate into a single, industry-wide, regulator-recognized standard. In practice, the role may vary substantially from one organization to another. In some studies, the medical monitor is deeply involved in protocol development, eligibility review, safety surveillance, adverse event assessment, protocol deviation review, medical data review, site escalation, investigator support, and DSMB or safety committee interactions. In others, the role is narrower, reactive, or insufficiently documented.
That variability creates risk. Not necessarily because some medical monitors lack expertise, but because the industry lacks a consistent way to define, verify, and maintain the competencies required for the role. And sometimes medical monitors do lack the necessary expertise.
What the poll tells us
The poll results suggest that the industry supports medical monitor certification. A practical model could include a common baseline certificate, an advanced certificate for high-risk or complex trials, and optional therapeutic-area or modality modules.
The goal should not be to create another administrative hurdle or to imply that certification can replace clinical experience. Poorly designed certification programs would add cost without improving results. The value of a framework would depend on whether it is practical, case-based, risk-proportionate, and clearly tied to decisions medical monitors make during trial conduct.
What a practical framework could look like
A practical medical monitoring certification framework should be demonstrated around competency, judgment, and risk, not paperwork. A useful model could include three levels:
1. Foundational Medical Monitoring Certification
This would apply to physicians or qualified health-care professionals supporting clinical trials. Core competencies would include GCP, regulatory responsibilities, participant protection, informed consent process, safety surveillance, adverse event assessment, protocol interpretation, medical data review, documentation standards, and clearly defined escalation pathways.
2. Advanced Certification for High-Risk or Complex Trials
This level would be intended for medical monitors supporting studies with elevated medical, operational, or regulatory complexity, including first-in-human trials, complex oncology programs, rare disease studies, gene and cell therapy development, high-risk device investigations, adaptive or platform trial designs, and research involving vulnerable populations. Additional competencies could include dose-escalation governance, monitoring, pausing, and stopping criteria, emerging safety signal management, benefit-risk assessment, DSMB/Data Monitoring Committee (DMC) interface, blinded vs. unblinded safety management processes, and crisis escalation procedures.
3. Therapeutic Area or Modality Modules
Optional specialization modules could further strengthen this framework by addressing therapeutic areas and operational domains where medical monitoring demands distinct expertise such as oncology, immunology, infectious disease, hepatology, neurology/CNS, cardiovascular, endocrinology, dermatology, gastroenterology, rare disease, pediatrics, women’s health, medical devices, digital endpoints, advanced biologics, gene and cell therapies, real world evidence integration, decentralized trials, or advanced therapies. This would preserve flexibility while recognizing that medical monitoring expertise is not interchangeable across all contexts.
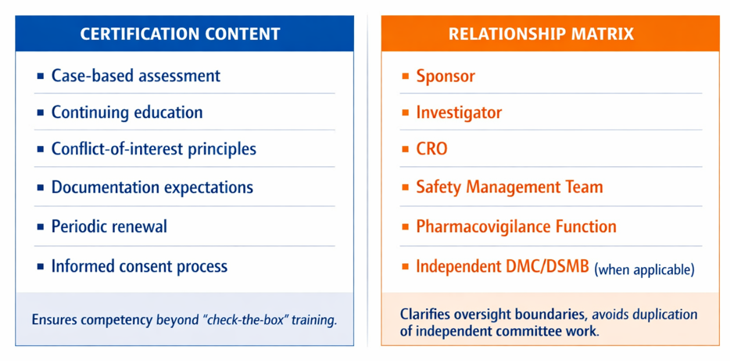
To avoid becoming a “check-the-box” exercise, certification should include case-based assessment, continuing education, conflict-of-interest principles, documentation expectations, and periodic renewal. It should also clarify the medical monitor’s relationship matrix regarding trial conduct with the sponsor, investigator, CRO, safety management team, pharmacovigilance function, and independent DMC/DSMB when applicable. FDA’s DMC guidance underscores the importance of determining when independent committees are useful and how their operation should be structured; a medical monitoring framework should complement, not duplicate, that oversight.⁷
To bring this idea to life, Table 2 below shows how certification can stay meaningful, grounded in real‑world case work and clear professional boundaries. It highlights the essential competencies and the relationships that define effective medical monitoring oversight.
Table 2: Medical Monitoring Certification Framework
The right framework would not replace sponsor accountability, therapeutic expertise, clinical judgment, or trial-specific training. It would create a common baseline for what “qualified” means in practice and provide a more transparent way to demonstrate readiness for a role that directly affects participant safety and trial integrity.
A practical and achievable initial step would not need to be a regulator-mandated credential. Instead, the industry could begin with a voluntary consensus-based competency framework developed collaboratively by key stakeholders including sponsors, CROs, academic clinical trialists, professional societies, experienced medical monitors, patient representatives, and regulatory bodies. Such an approach would allow for scalable standardization while preserving flexibility and stakeholder alignment. An initial pilot program could reasonably focus on high-risk interventional trials, where oversight demands are the greatest, competency gaps may carry the highest consequences, and the potential value of standardized medical monitoring qualifications is most immediately evident.
A call to action
The poll’s message is clear: the clinical research community sees a gap and is ready to discuss solutions.
Moving forward, progress will likely require a collaborative, multi-stakeholder approach. Sponsors, CROs, investigators, regulators, professional societies, academic medical centers, patient representatives, and experienced medical monitors should work together to define a practical competency framework. An initial pilot approach focused on high-risk trials, where the need is most obvious and the value proposition is strongest.
Medical monitoring has evolved well beyond a purely supportive role and now functions as a strategic safeguard for participant safety, protocol integrity, and risk management. As clinical trials continue to increase in complexity, reliance on inconsistent interpretations of this role may no longer be sufficient.
While a formal, risk-based certification framework would not eliminate sponsor accountability, clinical judgment, or trial-specific training, it could represent an important step toward consistency, transparency, and trust.
The question is no longer whether medical monitoring is essential; the question is whether we are ready to define the standard it deserves.
Disclaimer: This issue references an informal poll conducted on LinkedIn. LinkedIn did not sponsor, endorse, or participate in the poll. Content is for general informational purposes only.
Practical Pointer / May 2026
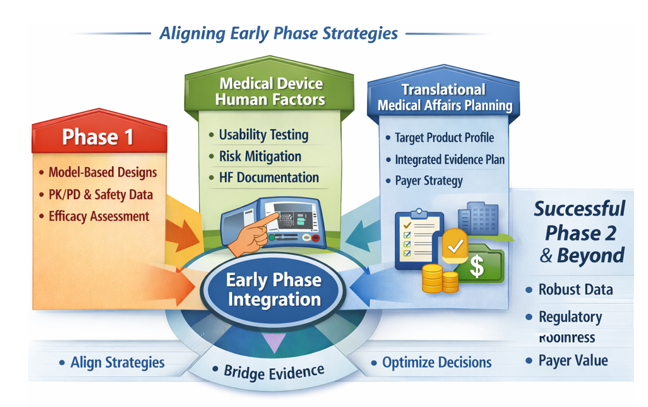
This practical pointer synthesizes three upstream development priorities: Oncology, rare disease, and complex immunology dose‑finding studies, Medical‑device human factors, and Translational Medical Affairs (TMA) evidence planning. This is incorporated into a single, actionable framework for sponsors preparing early‑phase protocols and regulatory submissions and want to get an early read on efficacy.
Phase 1 Oncology (as well as Rare Disease and Autoimmune) Clinical Trials: The 3+3 Default is no Longer Enough for Selecting a Dose
Modern early‑phase oncology development has shifted. The FDA’s January 2026 draft Bayesian guidance explicitly addresses oncology dose‑finding and notes that targeted therapies often require optimized dose selection, not simply identification of the maximum tolerated dose (MTD).[1] As a result, Bayesian and model‑assisted designs: CRM, BLRM, BOIN, mTPI/mTPI2, and efficacy‑toxicity designs are no longer academic alternatives; they are increasingly expected in early oncology programs.[2]
· CRM: Continual Reassessment Method: a model‑based design that continuously updates toxicity estimates to guide escalation more efficiently than rule‑based methods.
· BLRM: Bayesian Logistic Regression Model: a Bayesian dose‑toxicity model (often with escalation with overdose control (EWOC)) that provides flexible, probabilistic control of escalation decisions.
· BOIN: Bayesian Optimal Interval design: a model‑assisted design using optimized decision boundaries for escalate/stay/de‑escalate choices; simpler than CRM/BLRM but statistically stronger than 3+3.
· mTPI / mTPI‑2: Modified Toxicity Probability Interval: Bayesian interval‑based designs that classify doses as below, near, or above the target toxicity rate; mTPI‑2 improves decision consistency.
· Efficacy–toxicity designs: designs that jointly model both efficacy and toxicity to identify an optimal biological dose (OBD), not just the maximum tolerated dose (MTD).
A traditional 3+3 design begins by treating three patients at the starting dose and observing whether any dose‑limiting toxicities (DLTs) occur. Based on the number of DLTs, the next step is predetermined: escalate, expand to six patients, or stop and declare the previous dose the maximum tolerated dose. While familiar, this design provides limited information for modern agents and does not inseparably support dose optimization.
Early‑phase programs should justify dose‑escalation design rather than employing the traditional 3+3 model due to its perceived universal acceptance.
The FDAs new 2026 Bayesian draft guidance emphasizes:
Use of model‑assisted or model‑based designs
Inclusion of operating‑characteristic simulations
Clear articulation of how pharmacokinetic/pharmacodynamic (PK/PD), safety, tolerability, and early activity data will be integrated to guide dose selection
Consideration of more than one biologically active dose for optimization rather than defaulting to the highest tolerated dose
Critical Design Questions Before Protocol Finalization
Is the dose-escalation method justified, not simply inherited?
Do simulations demonstrate escalation/de-escalation behavior, target toxicity accuracy, and the probability of selecting an appropriate dose?
How will PK/PD, safety, tolerability, and early activity data be used to guide dose selection?
Is there a plan to evaluate more than one biologically active dose?
Practical Pointer: A traditional 3+3 design can still be proposed, but it should not appear to be the default because it is familiar. Sponsors should document why the selected design is fit for purpose, how it protects subjects, and how it will support Project Optimus; style dose optimization, which requires early, data‑driven evaluation of multiple active doses using PK/PD, exposure/ response, safety, and early activity data before registration trials.[3]
Medical Devices: Human Factors is Now an Upstream Regulatory Decision
The practical shift: Human factors (HF) refers to the systematic evaluation of how real users interact with a device, focusing on use‑related risks, critical tasks, and the potential for use errors that could lead to harm.
FDA’s final human factors guidance for medical device marketing submissions was issued in late May 2026 and establishes a risk-based framework for what human factors information belongs in 510(k), De Novo, premarket approval (PMA), and humanitarian device exemption (HDE) submissions.[1] The core decision is the HF Submission Category:
· Category 1 generally needs a conclusion and high-level summary
· Category 2 needs a rationale that no critical tasks or impacted critical tasks require validation data
· Category 3 needs a full Human Factors Engineering (HFE)/Usability Engineering (UE) report addressing validation testing for critical tasks
FDA also describes a 60-day operationalization window for submissions received before August 1, 2026.[5]
HF1: high-level summary
HF2: Rationale-based report
HF3: Full validation report
Translational Medical Affairs (TMA): Build the Integrated Evidence Plan Before Phase 2
The practical shift: The Integrated Evidence Plan should be treated as a living development-stage evidence roadmap, initiated no later than end-of-Phase 1/pre-Phase 2 and updated as clinical, regulatory, payer, and external stakeholder assumptions evolve. The IEP should map each decision-maker question (regulator, payer, prescriber, patient, and investigator) to a specific evidence source, endpoint, comparator, method, and timepoint.[6],[7]
Three linked planning documents that teams should evidence generation across development:[8] The TPP defines the intended product profile and claims; the CDP defines the clinical studies needed to support approval; the IEP defines the broader evidence needed to support regulatory, payer, clinician, patient, and post-approval decision-making.
The Target Product Profile (TPP) is a strategic planning tool that defines the intended efficacy, safety, and usability goals for the therapeutic under development. It outlines the key objectives the product must meet at each stage, supports go/no‑go decisions, and anchors development plans to the team’s discussions with regulatory agencies regarding evidence needs for submission.
The Clinical Development Plan (CDP) A plan which includes the overall development strategy, proposed phase 1, phase 2, and phase 3 study designs, dose-selection approach, endpoints, safety monitoring plan, regulatory strategy, statistical considerations, and major decision points. It also aligns clinical activities with nonclinical, CMC, pharmacovigilance, medical affairs, and commercial objectives.
The Integrated Evidence Plan (IEP) a cross‑functional plan that identifies the key questions decision makers will ask and the data needed to answer them. It outlines how clinical, regulatory, commercial, and patient‑ experience evidence will be generated, the timing and methods for producing that evidence within the clinical development plan, and how it will support both the regulatory submission and potential peri and post‑approval questions about real‑world use. The IEP should have explicit governance, with cross-functional input from Clinical Development, Regulatory, Medical Affairs, HEOR, Market Access, Patient Engagement, Biostatistics, Safety, and Medical Communications/Publications, and should be revisited at major data readouts, when new external data (e.g., competitor, regulatory, market) become available, and protocol decision points.
Use phase 2 to pre-position payer evidence
· Employ Disease‑specific patient-reported outcomes (PROs) and health‑utility measures: Use validated patient‑reported outcome tools tailored to the condition being studied, (as much as possible) along with standardized health‑utility instruments such as the SF-36 or EQ‑5D‑5L when relevant. These measures capture how patients feel and function in daily life, quantify symptoms and treatment burden directly from the patient perspective, and provide utility values needed for health‑economic analyses and value assessments.
· Collect healthcare-resource utilization and patient-burden data prospectively.
· Avoid relying solely on post-hoc indirect comparisons when feasible comparators could have been embedded earlier.[9]
Make field medical insights an evidence-planning asset
· Use structured insight capture tied to IEP evidence gaps, not free-text anecdote collection.
· Run a quarterly insights-to-evidence review with documented actions.
· Measure actioned insights, investigator-initiated study (IIS) opportunities, real-world evidence (RWE) analyses, and medical communications/publications, not just interaction counts.
Unified Practical Pointers for Sponsors
Before finalizing the next protocol or submission, sponsors should:
· Justify dose finding design with simulations and dose optimization logic
· Assign the HF submission category as part of the design controls and align with Quality Management system Regulation (QMSR)
· The output of the IEP should not be a static slide deck. It should be a decision tool that identifies which evidence gaps must be closed before phase 2, before pivotal trial initiation, before submission, and before launch.
Treat these as integrated development decisions, not isolated regulatory tasks. The strongest programs will align dose strategy, human-factors planning, and evidence generation early, reducing downstream risk and strengthening regulatory and payer readiness.
References:
[1] U.S. Food and Drug Administration. Use of Bayesian Methodology in Clinical Trials of Drugs and Biologics: Draft Guidance for Industry. Published January 12, 2026.
[2] Yuan J. A practical summary of FDA draft guidance: Use of Bayesian methodology in clinical trials of drug and biological products (January 2026). Published January 18, 2026.
[3] U.S. Food and Drug Administration. Project Optimus. FDA Oncology Center of Excellence. Accessed June 2026. https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus
[4] U.S. Food and Drug Administration. Content of Human Factors Information in Medical Device Marketing Submissions: Guidance for Industry and FDA Staff. Final Guidance. Published May 2026.
[5] U.S. Food and Drug Administration. Final guidance for human factors information in medical device marketing submissions. FDA Bulletin. Published May 28, 2026.
[6] Evidinno Outcomes Research. Integrated Evidence Generation Plans in Modern Biopharma. Published 2025.
[7] Lumanity. Integrated Evidence Generation Planning: From Strategy to Execution. Published 2026.
[8] Medical Affairs Professional Society (MAPS). Strategic Integrated Evidence Generation Planning: Company and Non‑Company Sponsored Research Guidance Document. Published 2024.
[9] Prescient Healthcare Group. Developing a Best‑in‑Class Integrated Evidence Plan: From Strategy to Studies. Published 2024.
Technology Innovation Spotlight - Reading Disease Signals from the Gut: An East Carolina University platform for fecal protein bioprofiling in inflammatory and neurological disorders

Methods and compositions for identifying fecal protein bioprofiles and treating inflammatory and neurological disorders
Technology Transfer Spotlight is a new MedSurgPI (MSPI) feature designed to translate academic innovation into clear industry opportunity. This first spotlight highlights an East Carolina University (ECU) invention published as WO2025155794A1, focused on subject-specific fecal protein bioprofiles, risk assessment, longitudinal monitoring, and kit-enabled use across inflammatory, neurological, neurodegenerative, and aging-associated disorders.
THE OPPORTUNITY: Inflammatory, neurological, and aging-associated disorders are biologically complex and often recognized only after meaningful disease progression. Developers and clinicians need biomarker approaches that are non-invasive, repeatable, and capable of capturing more than a single signal. ECU’s published application is directed at that gap by using stool as an accessible matrix for disease-related host-protein analysis.
THE INNOVATION: The invention establishes a subject-specific fecal protein bioprofile by extracting proteins from fecal material, determining relative abundance, grouping proteins by biological pathway, and generating a unique profile. The published application further describes using protein pathway enrichment scores to assess risk for inflammatory, neurological, neurodegenerative, and aging-associated disorders, and to monitor disease progression over time. The abstract also expressly includes kits for carrying out the method.
WHY IT MATTERS: First, it is non-invasive and well suited to serial sampling. Stool collection is better suited to repeated monitoring than many conventional approaches, which makes it attractive for patient-friendly screening and follow-up. Second, the signal is pathway-based rather than single-analyte based, creating room for broader biological interpretation, including immune activity, gut inflammation, and host-response patterns that may not be apparent from a single marker. Third, the same platform framework is positioned across inflammatory disorders, neurological disease, neurodegeneration, and aging-associated conditions an unusually broad span for one early translational asset.
TECHNOLOGY TRANSFER PERSPECTIVE: The intellectual property links sample collection, host-protein profiling, scoring logic, monitoring, and kitenabled deployment in one framework. That creates multiple partnering angles, including diagnostics, clinical biomarker strategy, proteomics service models, and remote disease-monitoring workflows. Those commercial pathways are an inference from the profiling, kit, and dashboard concepts described in the published application.
STRATEGIC INSIGHT: Many biomarker programs solve only for signal generation. This ECU asset addresses how a real-world workflow could function: collect the sample, isolate the relevant signal, interpret the biology, and potentially report changes over time through a usable platform. That combination supports positioning as a scalable translational platform rather than only a discovery-stage assay.
CALL TO ACTION: For additional information or partnership discussions, contact the ECU Office of Licensing & Commercialization. ECU contact Carlyle Rogers, Director of Licensing and Commercialization, innovation@ecu.edu.
PRACTICAL POINTER: The platform value of an academic invention increases when the IP covers not just the biomarker signal, but also the collection method, scoring framework, and deployment model.
Technology Snapshot
Institution: East Carolina University
IP Asset: WO2025155794A1 | WIPO (PCT) PCT/US2025/011987
Status/Dates: Published Jul 24, 2025 | Priority Jan 17, 2024 | Filed Jan 17, 2025
Inventors: Tonya Nicole Zeczycki, Stefan Clemens, Whitney Gabrielle Bond
Modality: Stool-based host-protein bioprofiling
Use Cases: Risk assessment, monitoring, kit-enabled testing
Classification: Go1N33/68
Partner Fit: Diagnostics, proteomics, digital health, CRO / biomarker partners

Free Cash Flow: What It Is and Why It Matters for Biotech and Other Startups

Authors: Gerald L. Klein, MD1 and Alex Walden, MBA1; Larry Florin, MBA2; Rob Walsh, MD1
Affiliations: MedSurgPI, LLC1; LBF BioPharma Consulting2
For early-stage companies, cash is not just a balance sheet line. It is time, leverage, and strategic optionality. It pays clinical trial invoices, keeps the team moving, and gives leadership room to make decisions before the market makes them instead. Profit on paper does not pay salaries, CRO invoices, regulatory consultants, server bills, or vendors. Free cash flow does.
Free cash flow, often abbreviated FCF, is one of the clearest measures of whether a business is generating more cash than it consumes. It cuts through accounting noise and focuses on the practical question every founder, board member, investor, and operating team eventually has to answer: after running the business and funding required investment, how much cash is actually left?
The standard formula is simple:
FCF = Cash Flow from Operations - Capital Expenditures
Cash flow from operations starts with net income and adjusts for non-cash items and working-capital changes. Capital expenditures (CapEx) include spending on equipment, facilities, laboratory infrastructure, manufacturing assets, technology systems, and capitalized software. Subtracting CapEx gives leadership a more realistic picture of available cash.
Many startups report negative FCF for years. That is not automatically bad. A biotech company advancing a promising therapeutic, device, diagnostic, or platform may appropriately consume cash before revenue begins. The real issue is whether negative FCF is intentional, milestone-driven, and adequately funded.
Why FCF matters more than “profit on paper”
Traditional profitability metrics can be useful, but they often miss what matters most in early-stage companies. Net income may include accounting treatments that do not reflect immediate cash movement. Earnings Before Interest, Taxes, Depreciation, and Amortization (EBITDA) can make a company look stronger by excluding items that still affect cash needs. Revenue growth can look impressive while the underlying business still consumes cash.
FCF brings the conversation back to operating reality. It asks whether the company has enough cash to execute the plan, reach the next milestone, and preserve negotiating power.
1. FCF defines runway
Runway is one of the most important numbers in any startup boardroom. A company with $36 million in cash and a monthly cash burn of $3 million has approximately 12 months of runway. That number drives hiring plans, trial design, vendor negotiations, fundraising timing, partnership outreach, and whether a company can wait for stronger data before raising capital.
In biotech and medtech, runway should not be modeled as one flat average. Cash burn changes by development stage. Preclinical work may be modest. Phase 1 and phase 2 trials increase costs through sites, monitoring, data management, safety oversight, and Clinical Research Organization (CRO) support. Phase 3 trials, manufacturing scale-up, regulatory filings, and launch preparation can change the cash profile dramatically. The organization’s Financial Planning and Analysis (FP&A) function needs to consider the timing of the future cash outflows and inflows (milestone payments). To the above example, that might mean a company with $36 million in cash may only have 9 months left because of known payments. Conversely, said company may hit a milestone in seven months and end up with an additional 18 months of cash.
2. FCF exposes the real cost of growth
Growth is attractive only when it is financially sustainable. A Software as a Service (SaaS) company can grow revenue while spending aggressively on customer acquisition. A medtech company can expand evidence generation while absorbing device manufacturing, quality-systems, and regulatory costs. A biotech company can advance a pipeline while committing to expensive clinical, chemical, manufacturing, and controls (CMC), pharmacovigilance, and medical affairs work.
FCF forces leadership to connect scientific and commercial progress with financial reality. A milestone is not only a scientific event. It is also a cash event. The question is not just, “Can we get to the readout?” It is, “Can we get to the readout with enough cash and credibility to act on the result?”
3. FCF is the language of capital
Investors do not fund narratives alone. They fund risk reduction, value creation, and credible use of proceeds. Venture investors, crossover funds, strategic partners, lenders, and acquirers all want to understand how cash will be deployed and what milestone changes the company’s financing profile.
For biotech companies, this often means modeling cash burn through commercialization. While accomplishing the next milestone will be the priority of the organization, losing track of the long FCF projection can become detrimental. Not properly forecasting FCF is inefficient in the best of cases. Biotech history is filled with companies that advanced promising science but exhausted available capital before development milestones could create financing leverage. In many cases, the constraint to success was not scientific viability alone, but the inability to sustain free cash flow through increasingly expensive stages of development.
A management team that understands FCF can explain which expenditures are milestone-critical, which are discretionary, which can be delayed, and which create leverage in financing or partnering discussions. That clarity can improve fundraising conversations and reduce surprises.
4. FCF signals operating discipline
In strong capital markets, inefficient spending can hide behind optimism. In tighter markets, it becomes visible quickly. Boards and investors scrutinize burn, hiring, vendor commitments, consulting spend, and capital expenditures. Managing FCF well means aligning spend with value creation. Periodically reviewing the assumptions surrounding the FCF projection is prudent. This process may highlight changes in the assumptions or the market or both. In that case, the company will have plenty of time to take corrective actions. Those actions may include finding a development partner at favorable terms.
That may include negotiating better supplier terms, sequencing trials intelligently, avoiding premature fixed costs, using fractional leadership, reassessing non-core programs, and timing CapEx around milestones.
The MedSurgPI Business & Financial Insights Lens
Biotech FCF requires a specialized view. R&D is often the dominant cash outflow. Clinical trials, CRO contracts, medical monitoring, safety oversight, manufacturing, regulatory preparation, scientific communications, and medical affairs support may appear as operating expenses, but they behave like strategic investments.
One-time inflows can also distort the picture. Licensing fees, grants, partnership payments, and milestone receipts may create a positive FCF quarter while the underlying business still burns cash. That is why leadership should separate recurring operating cash flow from one-time inflows and maintain a clear “core burn” view.
A practical approach is to forecast FCF by milestone: preclinical package, IND submission, phase 1 completion, proof-of-concept readout, pivotal planning, approval, and launch preparation. Each stage should have its own assumptions, risk points, financing requirements, and decision gates. A seasoned CFO will consider all the events and their likelihood and develop a cash projection.
The bottom line
Free cash flow is not a finance-department exercise or a year-end cleanup metric. It is the operational heartbeat of a startup. For biotech, medtech, and other innovation-driven companies, it determines how long the company can pursue its mission, how much flexibility management has, and whether the next strategic decision is made by the company or imposed by the market.
Companies that master FCF can choose when to raise, when to partner, when to expand, and when to conserve. Companies that do not often discover too late that progress without cash discipline is not enough and potentially fatal to the organization. A start-up biotech CFO will have a good handle on the assumptions surrounding the FCF and most importantly, will track changes to those assumptions.
MedSurgPI Business & Financial Insights will continue to explore practical financial, operational, and development topics that help founders, boards, investors, and clinical leaders make better decisions in capital-constrained environments.
Disclaimer: This article is for educational and informational purposes only and does not constitute financial, investment, accounting, tax, legal, regulatory, or medical advice. Readers should consult appropriate professional advisors before making business, financial, investment, or regulatory decisions.
Practical Pointers for April 2026
Authors: Gerald L. Klein, MD[1]; Freddy Byrth[1]; Roger E. Morgan, MD[1]; Michael Fath, PhD[2]; Patrick Loebs[1]; Shabnam Vaezzadeh, MD]3]; Larry Florin[4]; Kenny Carberry[5]; Eric Hacher[l], PhD6; Shengjun Zhang, MD[7]
Affiliations: MedSurgPI[1]; Cavabio Consulting[2]; Exquisite Biomedical Consulting[3]; LBF Biomedical Consulting[4]; Carberry Clinical Consulting[5]; SMART BioWorks, LLC[6]; BiotrialMed[7]

Drugs, Devices, and Diagnostics
Operational and regulatory takeaways for development teams
Drugs
Hospitals, universities, and other institutions remain a large, underutilized source of ideas for new therapeutic products.[1] These organizations should encourage entrepreneurial activity by providing education, training, and incentives that help staff identify inventions and innovations, develop intellectual property, and prepare commercially viable development plans. They should also strengthen educational links between academicians and industry professionals, enabling both groups to better understand the challenges and advantages of each environment and to bridge gaps that slow progress.
Conducting clinical trials is long, risky, and expensive. The time it takes many academic institutions to negotiate contracts and receive Institutional Review Board (IRB) approval is often excessive.[2] Regarding clinical trial agreements, institutions must standardize them across the institution. Consider operational fixes: using master CTA templates, fallback Intellectual Property (IP) terms, predefined startup-friendly license terms, and escalation timelines for contract impasse. Sponsors spend extra time adjusting to differing templates from one therapy area to the next within institutions, which exacerbates any review and revision time within the institution. This not only delays enrollment on the study but may shift sponsors and Contract Research Organizations (CROs) away from academics and more towards smaller, more agile centers. Standard agreements, such as Accelerated Clinical Trial Agreement (ACDA) and Accelerated Clinical Trial Agreement (ACTA) developed by major universities in collaboration with biopharma organizations and CROs have proven effective but remain underused. Likewise, use of the Streamlined, Multisite, Accelerated Resources for Trials Institutional Review Board (SMART IRB) Reliance model can reduce significant amounts of time associated with the IRB submission and approval process. Institutions should revise their technology transfer procedures to eliminate unnecessary bureaucracy and streamline operations. In particular, IP licensing or assignment terms should account for the financial and timeline risks that are being assumed by a startup entity intent on commercializing drug therapies. With current funding pressures, there is added urgency to adopt these efficiencies now. Doing so would increase productivity and strengthen collaboration between industry and academia.
Let us know your thoughts: info@medsurgpi.com
Devices
FDA’s March 2026 final guidance on Patient Preference Information (PPI) can be useful across the total product lifecycle for certain devices in several major ways, including:[1]
● Identifying unmet device needs and potential treatment options
● Determining endpoint prioritization or selection
● Determining effectiveness targets for clinical studies
● Informing clinically meaningful change in an endpoint
● Helping identify the most important benefits and risks from a patient’s and caregiver’s perspective
● Assessing the relative importance to patients of different benefit-risk attributes
● Clarifying how patients think about tradeoffs between benefits and risks
● Understanding heterogeneity in patient preferences, including subgroup considerations for benefit-risk assessments.[2]
How to know if your new device fits a 510(k) pathway
Start with the predicate-device question:
● Is there an FDA-cleared predicate device[3] with:
The same intended use, and
The same technological characteristics, or different ones that do not raise new questions of safety or effectiveness?
o A 501(k) is not merely a predicate search exercise. The sponsor must support substantial
equivalence with performance data when technical differences exist.
Ways to answer this include:
● Searching the FDA 510(k) database.
● Searching the classification database to see how FDA classifies similar devices.
● Submitting a briefing package through the FDA Pre-Submission (Pre-Sub/Q-Sub) program.[4] The FDA will respond in writing. This step should be grounded in a clear understanding of the numerous FDA and European Medicines Agency’s role within the EU Medical Device Regulation (EMA-MDR) guidance documents and directives that shape device development, including the critical requirement for a robust Quality Management System.
● Submitting a request for designation under 513(g). While this pathway provides a binding FDA determination of a product’s regulatory classification, it offers limited opportunity for dialogue or relationship-building with the Agency. For that reason, it is generally less advantageous than engaging through the Q-Sub process, which supports interactive feedback and more collaborative communication.[5]
Diagnostics
One of the biggest challenges in shipping blood specimens to central laboratories during clinical trials is temperature excursions and mechanical stress that cause sample degradation.[6],[7] A 2019 study found that 10% of red blood cell samples fell outside standard ranges, underscoring how vulnerable specimens are during transport.[8] Unfortunately, no harmonized guidelines exist for clinical-research-grade shipments.
Relevant resources include:
● WHO guidelines on the safety of shipping blood, and
● International Council for Standardization in Haematology (ICSH) materials, which address SOPs for specific analyses.
● Instructions from individual central laboratories, including validated packaging and handling requirements
Any labs or individuals with experience are invited to comment at info@medsurgpi.com.
Translational Medical Affairs (TMA)
Where medical insight becomes momentum: from first concept to lasting market impact
Patient education and training, and their understanding of how the pharmaceutical product relates to their individual health factors, are evolving rapidly. A patient’s and caregiver’s grasp of the importance and relevance of a treatment is essential for medication adherence. Written, audio, and video materials when combined with provider explanation, all contribute to better outcomes, especially when countering misinformation from unreliable online sources.[9]
Translational Medical Affairs (TMA) serves as the scientific bridge between early research, clinical development, and real-world medical practice, ensuring that insights from each stage inform the others.[10] Early engagement with clinicians and other health care professionals through TMA can surface questions about drug interactions, clinical benefits, and potential risks. Having this type of data in advance allows teams to frame answers and position the drug appropriately.[11]
Indications and claims for an approved drug depend entirely on the questions asked, endpoints selected, and data generated during clinical trials. This is often underappreciated. It is therefore critical that TMA help shape the right questions early so the development program yields the evidence needed for future regulator permitted claims.[12]

[1] Food and Drug Administration. Incorporating Voluntary Patient Preference Information Over the Total Product Life Cycle: Guidance for Industry, Food and Drug Administration Staff, and Other Interested Parties. March 2026. Published March 27, 2026. Accessed April 27, 2026.
[2] U.S. Food and Drug Administration. Patient Preference Information - Voluntary Submission, Review in PMAs, HDE Applications, and De Novo Requests, and Inclusion in Device Labeling. Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders. 2023.
[3] US Food and Drug Administration. The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]. Guidance for Industry and Food and Drug Administration Staff. FDA; July 28, 2014. Accessed April 27, 2026. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial-equivalence-premarket-notifications-510k
[4] Food and Drug Administration. Final Guidance on Incorporating Voluntary Patient Preference Information Over the Total Product Life Cycle. FDA Bulletin. March 27, 2026.
[5] Food and Drug Administration. How to Write a Request for Designation (RFD): Guidance for Industry. April 2011. Accessed May 7, 2026. FDA‑2011‑D‑0214.
[6] World Health Organization. Blood Cold Chain. WHO; 2011.
[7] International Council for Standardization in Haematology (ICSH). ICSH Recommendations for the Standardization of Haematology Review: Pre-analytical Variables and Sample Handling. Int J Lab Hematol. 2019;41(S1):1-29. doi:10.1111/ijlh.13011.
[8] Aalaei S, Amini S, Keramati MR, Shahraki H, Eslami S. Monitoring of Storage and Transportation Temperature Conditions in Red Blood Cell Units: A Cross-Sectional Study. Indian J Hematol Blood Transfus. 2019;35(2):304-312. doi:10.1007/s12288-018-1038-6
[9] Institute for Safe Medication Practices. Best Practices for Patient Education. ISMP; 2023.
[10] Klein G, Loebs P, Byrth F, Schacter L, Fath M. Translational Medical Affairs (TMA): A New Operating Model Bridging Preclinical Science, Clinical Development, and Real‑World Impact. SSRN. Published April 24, 2026. Accessed May 7, 2026. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=6640018
[11] American Medical Association. Principles of Translational Medical Affairs. AMA; 2022.
[12] US Food and Drug Administration. Labeling for Human Prescription Drug and Biological Products—Content and Format; Guidance for Industry. FDA; 2013.
[1] Kinch MS, Horn C, Kraft Z, Schwartz T. Rising academic contributions to drug development: evidence of vigor or trauma? ACS Pharmacol Transl Sci.
[2] Lawrence CE, Bruce V (Nickie) M, Salberg LD, et al. Quantitative assessment of the impact of standard agreement templates on multisite clinical trial start up time. Journal of Clinical and Translational Science. 2023;7(1):e204. doi:10.1017/cts.2023.622
Data and Safety Monitoring Board Best Practices in Clinical Trials

Abstract
A Data and Safety Monitoring Board (DSMB), also referred to as a Data Monitoring Committee (DMC), plays a critical role in safeguarding participant safety and ensuring the integrity of clinical trials. This article provides a narrative overview of best practices in DSMB structure, governance, and operations, informed by regulatory guidance from the U.S. Food and Drug Administration (FDA), National Institutes of Health (NIH), European Medicines Agency (EMA), and selected literature, as well as practical experience in clinical trial oversight. Key domains addressed include DSMB composition and independence, charter development and governance, data access and integrity, safety monitoring processes, interim analyses and adaptive decisionmaking, confidentiality, and sponsor responsibilities. The article also discusses common operational challenges and mitigation strategies, along with emerging trends such as Bayesian monitoring approaches and decentralized trial oversight. Rather than prescribing specific statistical thresholds, the manuscript emphasizes principles for maintaining scientific rigor, ethical oversight, and regulatory alignment. This overview is intended to support sponsors, Clinical Research Organizations (CROs), and DSMB members in implementing effective and compliant safety monitoring practices across a range of clinical trial settings.
Read the full manuscript in Cureus
Translational Medical Affairs (TMA): A New Operating Model Bridging Preclinical Science, Clinical Development, and Real-World Impact

Authors: Gerald L. Klein, MD; Patrick Loebs; Freddy Byrth; Lee Schacter, PhD, MD; Michael Fath, PhD
Introduction
Medical Affairs (MA) is undergoing a fundamental transformation. Historically positioned as a late-stage, support-oriented function focused on scientific exchange and data dissemination, MA is now expected to play a far more strategic role in evidence generation and decision-making.
The drivers are clear:
● Increasing regulatory complexity
● Growing reliance on real-world evidence (RWE)
● Heightened expectations from payers and Health Technology Assessment ( HTA) bodies
● Persistent evidence gaps at launch
● Rapid integration of AI and advanced analytics
Traditional MA models—largely reactive and downstream—are no longer sufficient. A new model is emerging: Translational Medical Affairs (TMA).
What Is Translational Medical Affairs?
Unlike traditional MA inclusion strategies, in which MA is engaged late in the development cycle, TMA refers to a clinical development operating model where MA is embedded from the earliest stages of development as an integrated, lifecycle-spanning part of the interdisciplinary development team that augments the connections across preclinical development, clinical trial design and execution, real-world evidence generation, market access and payer strategy.
Why Preclinical Integration Is Critical
The most important decisions in drug development occur before the first patient is enrolled, including mechanism of action (MoA) selection, translational biomarker strategy, patient population definition, and early safety characterization.
Another critical consideration, particularly in therapeutic areas with a high density of active development programs, is the availability of patients for clinical trials. This includes not only the size of the addressable patient population, but also the number of ongoing and anticipated studies that may compete for the same patients, potentially impacting feasibility, enrollment timelines, and overall trial success.
In addition, early clinician input is essential in shaping study design and protocol execution, including inclusion and exclusion criteria and overall operational feasibility in real-world clinical settings.
TMA embeds Medical Affairs early, enabling validation of clinical relevance, alignment with ICH E8(R1) fit-for-purpose study design, anticipation of payer evidence needs, and identification of evidence gaps before costly Phase 1/2 trials.
Regulatory Alignment: A Key Driver
TMA aligns with evolving global regulatory expectations:
● ICH E6(R3) – Quality by Design and proactive risk management
● ICH E8(R1) – Fit-for-purpose study design aligned with stakeholder needs
● FDA Real-World Evidence Framework
● FDA Project Optimus
● FDA Artificial Intelligence/Machine Learning (AI/ML) guidance (January 2025
● EMA Artificial Intelligence Workplan (2023–2028)
These frameworks reinforce that evidence must be prospectively aligned with decisions—not retrospectively adapted.
Operationalizing Translational Medical Affairs
● Embed MA in preclinical development (Target Product Profile (TPP), biomarkers, MoA relevance)
The target product profile should be tightly linked to the overall clinical development plan, with periodic cross-functional clinical review to ensure continued alignment with emerging data, regulatory expectations, and strategic objectives.
● Develop a lifecycle evidence plan (regulatory, clinical, payer, RWE)
This lifecycle evidence plan should not function as a standalone document, but rather as an integrated component of the broader Clinical Development Plan, ensuring that evidence generation is coordinated, continuously updated, and aligned with key development and decision milestones.
● Integrate real-world evidence early (feasibility, endpoints, external validity)
Evolve MSLs into insight generators and evidence strategists
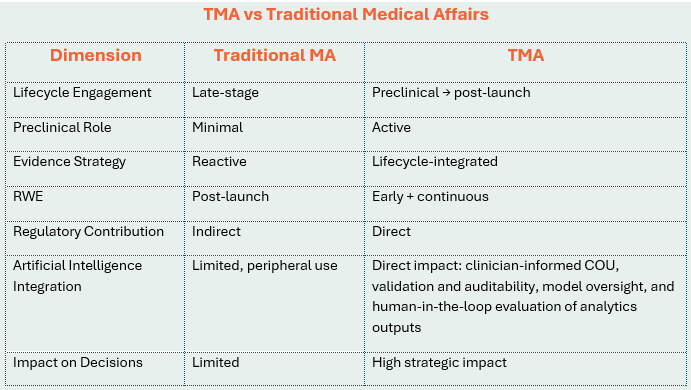
Strategic Value of TMA
● Higher probability of technical and regulatory success (PTRS)
● Reduced late-stage development risk
● Stronger payer positioning
● Faster clinical adoption
● More efficient resource utilization
● Generation of clinically meaningful insights to inform early prediction of risk-benefit profiles and logistical burden for both patients and clinical practice settings, including feasibility within routine care workflows.
The TMA Lifecycle Model
Preclinical → Clinical Development → Real-World Evidence → Market Access → Feedback to Development (continuous loop with bidirectional insight flow).
Conclusion
Translational Medical Affairs is a redefinition of the usual Medical Affairs function/construct. By integrating preclinical insight, clinical development, and real-world evidence, and the integration of artificial intelligence models and model development, TMA positions Medical Affairs as a strategic driver of development success, regulatory alignment, and clinical impact.
References
1. International Council for Harmonisation (ICH). E6(R3) Good Clinical Practice. 2025.
2. International Council for Harmonisation (ICH). E8(R1) General Considerations for Clinical Studies. 2021.
3. U.S. Food and Drug Administration. Framework for FDA’s Real-World Evidence Program. 2018.
4. U.S. Food and Drug Administration. Project Optimus. 2023.
5. European Medicines Agency. Artificial Intelligence Workplan 2023–2028.
6. U.S. Food and Drug Administration. Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations. Published January 2025.
7. U.S. Food and Drug Administration, Health Canada, Medicines and Healthcare products Regulatory Agency. Good Machine Learning Practice for Medical Device Development: Guiding Principles. Published 2021.
8. Sherman RE, Anderson SA, Dal Pan GJ, et al. Real-world evidence—what is it and what can it tell us? N Engl J Med. 2016;375(23):2293-2297.
9. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273-286.
10. Drummond MF, Schwartz JS, Jönsson B, et al. Key principles for the improved conduct of health technology assessments for resource allocation decisions. Nat Rev Drug Discov. 2008;7(3):255-263.
Practical Pointers for Drug Development and Translational Medical Affairs / March 2026

Authors: Gerald L. Klein, MD (1); Lee Schacter, PhD, MD (1); Larry Florin (2); Marion Stamp-Cole( 3); Patrick Loebs (1); Roger Morgan, MD (1); Emilia Jones Amaowei (4); Roger Nolan, PhD (1); Freddy Byrth (1)
Affiliations: MedSurgPI (1); LBF BioPharma Consulting (2); MSC Clinical Consulting (3); EJ MedInsights(4)
Drug Development
This paper examines how evolving FDA safety surveillance, data quality expectations, and participation challenges are reshaping clinical development and outlines the strategic role that Translational Medical Affairs (TMA) can play in addressing these shifts.
● The U.S. Food and Drug Administration launched the Adverse Event Monitoring System (AEMS) in March 2026 to “consolidate multiple disparate reporting systems currently used across all FDA-regulated product categories, including medical products, vaccines, devices, tobacco, food, cosmetics, and veterinary medicines.”[1] It is meant to do the following:
o Enhance data quality and consistency through standardized reporting protocols, streamline reporting processes to reduce administrative burden on both internal FDA staff and external stakeholders, and strengthen safety surveillance capabilities through advanced case processing workflows, AI-based redaction and digitization tools, enhanced analytics, and comprehensive cross-product surveillance.
o Serve as a centralized platform for consumer complaints, regulatory misconduct reports, and whistleblower submissions across all FDA centers.[2]
AEMS is also expected to enable convergence of traditionally distinct safety data streams, including solicited data collected in clinical trials and spontaneous post-marketing reports, thereby supporting a continuous, lifecycle-based safety surveillance framework from development through commercialization. In parallel, evolving expectations regarding “data fit for regulatory purpose” and the ongoing implementation of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use E19 guideline on selective safety data collection reflect a broader shift toward more targeted, risk-based data acquisition strategies. These developments align with the capabilities of AEMS, including the potential for near–real-time (e.g., daily) safety data ingestion and analysis early in the clinical development process.
● The U.S. Food and Drug Administration defines “data fit for regulatory purpose” as data that are relevant to the specific clinical and regulatory question and are sufficiently reliable, complete, and accurate to support regulatory decision-making; this concept is foundational to Quality by Design, requiring early identification and prospective incorporation of Critical to Quality (CtQ) factors to ensure data integrity from the outset of the study.[3] Clean data alone are not enough, data must also originate from fit-for-purpose endpoints and study designs aligned to the intended decision. If the data cannot directly support a regulatory or clinical decision, they are operationally costly, but strategically weak. Designing trials with decision-ready data in mind is now a competitive advantage. This raises several fundamental questions that should be addressed prospectively in study design:
o Is it relevant to the clinical and regulatory question?
o Is it reliable in how it was collected?
o Is it interpretable in the context of decision-making (regulatory, clinical, or reimbursement)?
o Is it free from systematic bias or adequately controlled to support valid inference?
● Participant enrollment and retention is directly proportional to clinical trial burden that is placed on these patients.[4] Enrollment and retention are influenced by a matrix of factors, including disease state and severity, the number and type of assessments (particular those that are invasive) and the evolving risk-benefit ratio as communicated to participants over the course of the study. Additional contributors include, among others, the travel distance, visit frequency and duration, procedural complexity, and the associated physical, psychological, and logistical burden (e.g., discomfort, time commitment) of study participation. This clinical trial burden in its entirety should be taken into consideration when estimating both the enrollment and retention rate of the study.
Translational Medical Affairs
● It is estimated that only about 3% of physicians in the United States actively participate in clinical trials.[1] Limited awareness of opportunities, uncertainty regarding regulatory requirements, and the perceived operational burden of trial participation all contribute to low engagement. As a result, enrollment capacity becomes a structural constraint in clinical development. Efforts are underway to address these barriers to physician participation through practical solutions such as patient concierge services, simplification of study materials (including informed consent forms), hybrid and decentralized study designs incorporating remote patient data capture, and expanded use of telehealth; however, while promising, these approaches remain early in adoption and there is still a long way to go before they meaningfully resolve these structural challenges. In this context, Medical Science Liaisons (MSLs) are uniquely positioned to address the gap by reframing clinical trial participation as a scientific and institutional opportunity, rather than an operational burden, thereby enhancing physician engagement and supporting more efficient study execution. MSLs can strategically position participation by emphasizing the following value drivers:
o Contribute advancing standards of care
o Gain early access to innovative therapies
o Access academic and publication opportunities
o Strengthen institutional prestige and patient retention
o Increase practice revenue
o Drive professional growth and stimulation
● Translational Medical Affairs should play a more prominent role in scientific publication, as it operates at the critical interface between clinical practice, research, and real-world evidence generation. This unique positioning enables TMA to translate emerging data into clinically meaningful insights that advance patient care and inform clinical, payer, and policy decision-making. Publications provide a systematic capture and disseminate insights derived from clinical trials, investigator interactions, and real-world experience, areas that are often underrepresented in traditional academic outputs. Increased publishing strengthens scientific credibility, closes evidence gaps not addressed by registrational studies, and reinforces the non-promotional value as a trusted independent scientific partner.[2] Moreover, a robust publication strategy enhances engagement with key opinion leaders, contributes to guideline development, and elevates organizational visibility in an increasingly competitive and data-driven healthcare environment.
● Translational Medical Affairs can play an important role, as part of the interdisciplinary clinical development team, in early phase 2 trials by initiating early external engagement with key opinion leaders to validate assumptions, gather actionable feedback, and build scientific credibility. It also identifies evidence gaps that inform subsequent study design. This approach ensures that phase 2 programs remain scientifically robust, clinically relevant, and strategically aligned for later-phase success.[3] As clinical development becomes increasingly data-driven, patient-centric, and complex, TMA must evolve from a supportive function to a strategic driver of early evidence generation, leveraging proactive stakeholder engagement, identification of evidence gaps, and translational insight to shape development programs and de-risk later-phase outcomes.
In this context, evolving regulatory expectations around “data fit for regulatory purpose,” coupled with initiatives such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use E19 guideline on selective safety data collection, further reinforce the need for targeted, high-quality data generation strategies. These trends align with emerging capabilities such as the FDA’s Adverse Event Monitoring System (AEMS), which is expected to support near–real-time safety data acquisition and to converge traditionally separate solicited clinical trial data and spontaneous post-marketing reports into a unified, lifecycle-based safety surveillance framework from development through commercialization.
[1] U.S. Food and Drug Administration. FDA Adverse Event Monitoring System (AEMS). Accessed March 31, 2026. https://www.fda.gov/drugs/surveillance/fda-adverse-event-monitoring-system-aems
[2] Ibid
[3] International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). E6(R3) Guideline for Good Clinical Practice. Step 4. 2025. Accessed March 31, 2026. https://www.ich.org
[4] Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185–198.
[5] Henry TA. Trial-in-a-box to help more practices take part in clinical trials. American Medical Association. Published May 27, 2021. Available at: https://www.ama-assn.org/practice-management/digital-health/trial-box-help-more-practices-take-part-clinical-trials. Accessed March 31, 2026.
[6] Wager E, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461-464.
[7] Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000-2009. Clin Pharmacol Ther. 2011;89(2):183-188.
Practical Pointers for Product Development and Medical Affairs / December 2025
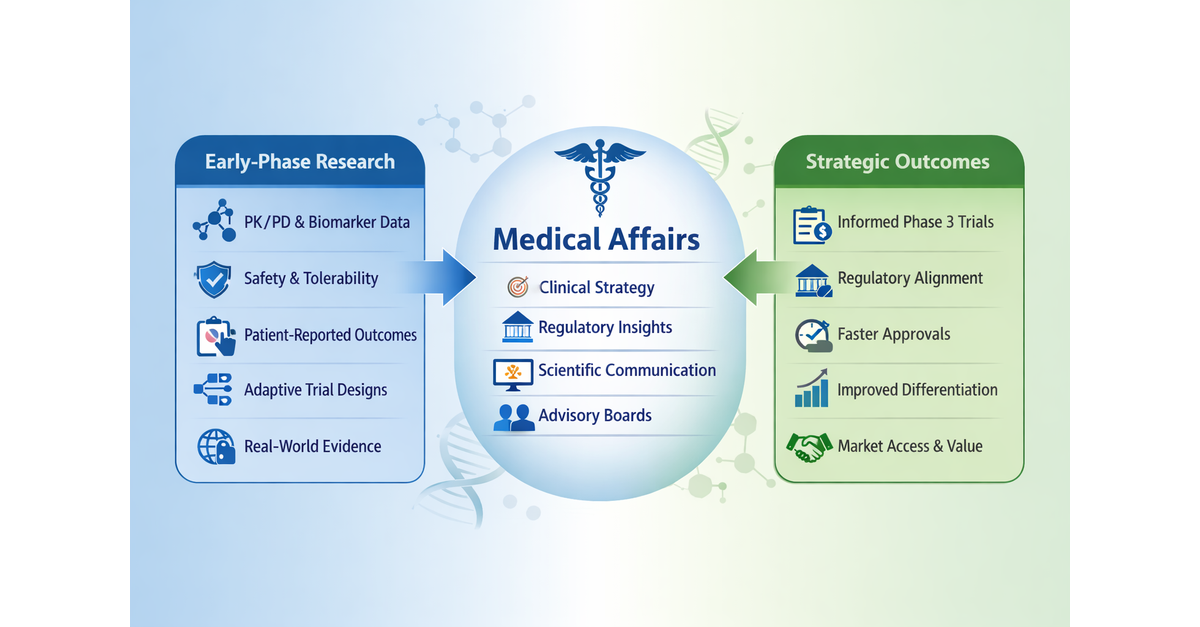
Practical Pointers for Drug Development / December 2025
Authors: Gerald L. Klein, MD1; Roger E. Morgan, MD1; Melissa Palmer, MD2; Freddy Byrth1; Michael Fath, PhD3; Katie-Louise Dawson, MD1; Colin Scott, MD4; Patrick Loebs1; Rob Walsh, MD1
Affiliations: MedSurgPI; Liver Consulting, LLC2; Cavabio Consulting3; Highland Airs Consulting4
Drug Development
Early-phase studies now play a larger role for generating clinical, mechanistic, and real-world insights that historically emerged only during phase 3 development.[1] Traditionally, phase 1 studies focused primarily on safety, tolerability, and pharmacokinetics, often conducted in healthy volunteers when appropriate. By incorporating elements relevant to future marketing applications, such as those required for New Drug Application (NDA), Biologics License Application (BLA), or Marketing Authorization Application (MAA) submissions, into phase 1 and phase 2 studies, development teams can generate critical evidence earlier in the program. [1,2] These elements may include response relationships, expanded safety datasets, patient-experience measures, and real-world clinical context. Early integration of these components helps prevent late-stage trials from becoming burdened with variables introduced too late in development, while strengthening alignment with the Target Product Profile (TPP) and the overall Clinical Drug Development Plan. [3]
Phase 1 trials should therefore be designed to provide maximum strategic value, reduce phase 2 failure risk, and strengthen investor and regulatory confidence [1,2]
o Although primarily intended for first-in-human safety and tolerability assessments, phase 1 trials should also function as value-creation platforms that:
§ Incorporate pharmacokinetics (PK) and pharmacodynamics (PD) modeling, when financially and otherwise feasible, to inform dose selection and variability assessment [3]
§ Identify exposure–response relationships early in order to inform phase 2 design[3]
§ Explore multiple dose levels with sufficient sampling density to characterize pharmacologic activity and, for targeted therapies such as oncology or immunomodulatory agents, define the biologically effective or optimal dose.
· Incorporating adaptive or model-informed approaches (including machine learning methods) to accelerate dose optimization and minimize exposure to subtherapeutic dosing. Examples include: [3,4]
o Bayesian dose-escalation designs [4]
o Model-informed drug development (MIDD) strategies aligned with FDA guidance [3]
o Real-time PK analysis for data-driven decision making [3]
o Adaptive cohort expansion to refine dose and population selection4
Importantly, the traditional use of healthy volunteers in phase 1 studies remains appropriate for many therapeutic classes, particularly when the anticipated safety profile permits such evaluation. Healthy volunteer studies allow clearer pharmacokinetic characterization, minimize confounding effects from underlying disease, and provide efficient assessment of tolerability and dose-related safety. This approach remains common for agents such as antihypertensives, antibiotics, metabolic drugs, and many neurological therapies.
However, in some therapeutic areas (oncology, gene therapies, immunotherapies, and other targeted or higher-risk modalities) phase 1 studies are frequently conducted directly in patients because of safety considerations, disease specificity, or the need to observe early signals of biological activity.
Hybrid early-phase designs are increasingly used, in which initial cohorts of healthy volunteers are evaluated for safety and pharmacokinetics, followed by patient cohorts that explore dose escalation, pharmacodynamic effects, and early signals of activity.
· In oncology, rare disease, or high-unmet-need indications, early efficacy-generating components may also be strategically integrated to support expedited development pathways, including: [5,6]
o Expansion cohorts within phase 1 protocols
o Early proof-of-concept arms
o Enrichment strategies (e.g., biomarker-selected populations) to increase the probability of demonstrating a meaningful signal
o Independent medical monitoring should be incorporated into all clinical studies involving human participants so that adequate safety is provided for the trial
· For immunomodulators, gene therapies, biologics, and other first-in-class agents, embed robust safety governance mechanisms consistent with ICH E6(R3) Quality-by-Design principles.
· These mechanisms may include:
o Independent Adjudicators
o Independent Safety Review Committee (SRC)
o Predefined halting/stopping rules, both for individual patients and the clinical trial
o Sentinel dosing for initial human exposure [7]
Medical Affairs
Medical Affairs should play a key role in early drug development by helping translate emerging clinical data into scientific, regulatory, and strategic insights. Early engagement supports cross-functional collaboration and the development of a robust TPP that guides clinical design, regulatory planning, and future product positioning. [8]
· Early engagement of Medical Affairs, alongside Regulatory Affairs, helps ensure that emerging clinical data are interpreted and positioned within the broader scientific and clinical context. [9-11] Areas where cross-functional planning is particularly important include:
o Evaluating special-population considerations (renal or hepatic impairment studies) [12]
o Assessing drug-drug interaction potential and food-effect impact proactively [12]
o Conducting QT assessment when relevant (ICH E14 alignment) [13]
o Evaluating immunogenicity risk for biologics and complex modalities [14]
· Medical Affairs also contributes to scientific and clinical advisory board strategy and asset differentiation by engaging internal and external subject matter experts (SMEs) [8-10] including:
o Identifying and engaging appropriate scientific advisory boards
o Helping develop key differentiators such as:
§ Positioning quality-of-life (QoL) endpoints and patient-reported outcomes (PROs) within the broader clinical and value narrative once study results are available[15]
§ Outcomes demonstrating advantages over current therapies
§ Regulatory credibility through alignment with FDA and global agency expectations[16]
§ Risk-reduction strategies that enhance program resilience
§ Strategies that increase asset valuation and partnership appeal
· Development teams should ensure early cross-functional collaboration and accountability to support the creation and refinement of a rigorous TPP.11 Key elements to consider include:
o Incorporating real-world evidence (e.g., observational studies, registries, or healthcare data sources) and early clinical insights from clinical practice and investigators
o Defining the proposed indication
o Specifying the dose and dosing regimen
o Clarifying the route of administration
o Defining the target population
o Anticipating the safety profile
o Articulating differentiation vs. standard of care
o Defining differentiation from competitive products
[1] Morgan P, et al. Impact of a five-dimensional framework on R&D productivity. Nat Rev Drug Discov.2018;17(3):167-181.
[2] Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61(3):275-291.
[3] US Food and Drug Administration. Target Product Profile—A Strategic Development Process Tool. 2007.
[4] Neuenschwander B, Branson M, Gsponer T. Bayesian adaptive dose-escalation designs. Stat Med. 2008;27(13):2420-2439.
[5] Beaver JA, Howie LJ, Pelosof L, et al. A 25-year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics. JAMA Oncol. 2018;4(6):849-856.
[6] US Food and Drug Administration. Expedited Programs for Serious Conditions—Drugs and Biologics: Guidance for Industry. FDA; 2014.
[7] US Food and Drug Administration. Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials. FDA; 2007.
[8] US Food and Drug Administration. Target Product Profile—A Strategic Development Process Tool. FDA; 2007.
[9] O’Connor D, Lee S, et al. The evolving role of medical affairs. Ther Innov Regul Sci. 2019;53(4):447-455.
[10] Engelberg AB, Kesselheim AS. Medical affairs and the shifting pharmaceutical landscape. N Engl J Med. 2016;374:1787-1789.
[11] Garattini S, Padula A, et al. The role of medical affairs in evidence generation. Lancet. 2020;395:1968-1970.
[12] US Food and Drug Administration. Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications: Guidance for Industry. FDA; 2020.
[13] International Council for Harmonisation. E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. 2005. Updated Q&A 2017.
[14] US Food and Drug Administration. Clinical Pharmacology Considerations for Biologics: Guidance for Industry. FDA; 2019.
[15] Basch E, Bennett AV. Patient-reported outcomes in drug development. JAMA. 2017;318(2):197-198.
[16] International Council for Harmonisation. ICH Harmonised Guideline E6(R3): Good Clinical Practice. 2023.
The Target Product Profile and the Development Plan - Inextricably Intertwined

Authors: Lee Schacter, PhD, MD and Gerald L. Klein, MD
The development of a new chemical entity is guided by two documents; the Target Product Profile and the Development Plan. The first is a wish list, the second, a road map to those wishes and the pot of gold that awaits if they are achieved.
Whether it is a new compound “tossed over the fence” from discovery to clinical in Big Pharma or the precious creation of a biotechnology company, the challenge for the clinical group is to turn a chemical entity into a marketable product.
Step one is the wish list. If the new compound, which we will call FRED, works as well as possible based on preclinical models, what is the ideal labeled indication for use that can be approved? For example, if FRED has been designed to treat rheumatoid arthritis, the dream label could read “FRED is indicated for the initial treatment of rheumatoid arthritis in adult and pediatric patients.” But not all dreams come true and sometimes it is necessary to accept a second or third place finish such as “FRED is approved for the treatment of rheumatoid arthritis in adults after the failure of initial therapy” or even worse; “FRED is approved for the treatment of adults with rheumatoid arthritis refractory to all other available modalities.”
Now the “game is afoot.” Will management be flexible with the label? Is the worst possible label acceptable to management? Will it represent an adequate return on investment to justify further development? What if the worst possible label is first to market for a new class of agents and there is an opportunity to expand the label? What compounds, if any, with the same mechanism of action are in development, and at what stage? How will FRED be positioned? What will be its therapeutic niche? Will it be first-in-class agent, the best-in-class among similar compounds, or a me-too product, perhaps differentiated only by a more favorable pharmacokinetic profile that supports a lower price point? All these factors and more (Table 1) have to be considered in the Target Product Profile.
Table 1
Considerations for the Target Product Profile
Expected clinical activity
Therapeutic needs (current and projected) the product can address
Comparisons to existing agents; strengths, weaknesses, opportunities, and threats (SWOT analysis)
Route of delivery vs. existing agents
Cost of goods
Overall competitive environment including marketed products and those in development
The Development Plan takes the Target Product Profile for FRED and creates a path to making dreams come true. This plan is based on the science behind the new agent, the treatment options currently available for the target disease, along with those in development, and the projected cost of development. The Development Plan should represent the “value proposition” for FRED. While it aims to account for both the current and future competitive environment for FRED, it must remain a living document, modified as new data for both FRED and for other agents, both approved and in development, become available.
A major consideration is cost and time in development. New drug development spending is stratospheric. A recent paper[1] concludes “that the median… total R&D expense, reflecting all clinical trials and including adjustments for inflation, capitalization, and discontinued products, was approximately …. $708 million” in 2019. They also note that “…costs captured by patient-months comprise most total … R&D spending….” Similar findings were reported elsewhere, estimating that “the mean cost of the clinical phase per drug candidate was $117.4 million”, with approximately $27 million for phase 1, $34 million for phase 2, and $38 million for phase 3.[2]
Faced with these costs and the success rate of new compounds (approximately 10-20%)[3,4] the development plan has to be focused on the rapid efficient evaluation of any new compound. This requires carefully crafted go/no go criteria at each stage of clinical trials along with timelines that are aggressive but achievable. The endpoints must also be chosen to support the earliest possible registration.
Critical elements therefore include:
Define the regulatory strategy for the US, EU, and other territories
o Determine if the compound will be developed as a first-in-class, best-in-class, or “me too” agent and where it will fit in the treatment paradigm of the target disease
Meet with regulatory authorities to discuss the proposed development and registration strategy, obtain feedback on the registration pathway, and align the clinical and nonclinical development program with regulatory requirements and expectations. Define the data needed for go/no go decisions for each stage of development (phases 1-3).
o In a competitive environment for patients and investigators, design studies need to be attractive to both by minimizing patient and site burden, and by keeping protocols focused and as simple as possible.
Avoid scientific side shows or academic curiosities that are of interest but not on the critical path to regulatory approval while collecting essential data for next generation drugs.
Track therapeutic landscape in the target disease including compounds in development and emerging relevant science.
———————————————————————
[1] Mulcahy AW, et al. Use of Clinical Trial Characteristics to Estimate Costs of New Drug Development. JAMA Network Open. 2025;8:e24533275. doi:10.1001/jamanetworkopen.2024.53275
[2] Setkaya A, et al. Costs of drug development and research and development intensity in the US, 2000–2018. JAMA Network Open. 2024;7:e2415445. doi:10.1001/jamanetworkopen.2024.15445
[3] Yamaguchi S, et al. Success rates of new compound development. Clin Transl Sci. 2021;14(3):1113-1122. doi:10.1111/cts.12978
[4] Duxin S, et. al. Why 90% of clinical drug development fails and how to improve it? Acta Pharma Sin B 2022; 12(7): 3039-3062
Contract Research Organizations - Trust but Verify
Authors: Lee Schacter, PhD, MD; Gerald L. Klein, MD
A recent news article published in Science[1] highlights disturbing incidences of apparent maleficence by Clinical Research Organizations (CROs) in South Florida. The article stated that the biotech T3D Therapeutics found data generated by a CRO from an Alzheimer’s trial to be “medically impossible”. Issues ranged from treatment of patients without evidence of the disease to levels of the test agent in patients receiving placebo and led to the write off of a $35 million study. Challenging for any company but especially for a startup.
Other companies have been burned by CROs conducting Alzheimer’s trials including Annovis Bio and BioVie according to the article, again with the enrollment of inappropriate patients or brain scans that were copied and pasted. Big Pharma is not immune to the issue. Bristol-Myers Squibb was burnt by irregularities leading to exclusion of data in a trial of Cobenfy and needs to replace subjects. A study by Eli Lily may also be affected.
Malfeasance by CROs happens and sponsors must be careful not only in their choice of CRO but should have their studies monitored independently and often. It is not a good practice to give your clinical trial to a CRO and not be involved until it is completed. Independent consultants to help with the in-house medical and clinical operations is a cost-effective way to protect the study and the company. Reviews need to be unblinded and detailed, conducted by experienced individuals able to spot irregularities as they occur and recommend corrective action. If a CRO is also responsible for any critical elements such as the development plan, study design, statistical plan, electronic medical record creation, and maintenance; these too need to be carefully reviewed by outside experts, especially for a small company to assure they will meet the sponsor’s requirements and the regulators’ needs. The extra expense of these activities is more than justified by the risk of misconduct or suboptimal performance by the CRO.
Bottom line: when a CRO manages a trial, sponsors should trust—but always verify.
[1] Kaiser J. Alzheimer’s drug developers accuse clinical trial sites of faking data. Science. 2026;371(6524):112–115. Published January 8, 2026. doi:10.1126/science.adkXXXX
Practical Pointers for Drug Development and Medical Affairs / November 2025

Practical Pointers for Drug Development and Medical Affairs
November 2025
Authors: Gerald L. Klein, MD1; Roger E. Morgan, MD1; Melissa Palmer, MD2; Freddy Byrth1; Michael Fath, PhD3; Thomas Krol, MD1; Shabnam Vaezzadeh, MD4; Mark Tulchinskiy, MD5
Affiliations: MedSurgPI1; Liver Consulting, LLC2; Cavabio Consulting3; Exquisite Biomedical Consulting4; Worldwide Clinical Trials5
Drug Development
Improving Academic Research Site (ARS) or Academic Medical Center (AMC) Readiness for Industry-Sponsored Trials
Due to their access to specialized patient populations, multidisciplinary expertise, and advanced research infrastructure, academic institutions may have the opportunity to play a critical role in conducting clinical trials, particularly those involving rare diseases, underserved or vulnerable populations and complex study designs. However, an increasing number of academic sites are being excluded from industry-sponsored studies because of elevated institutional fees, excessive administrative complexity, and significant delays in executing required study documents, particularly lengthy IRB review and approval timelines.[1],[2] This problem is compounded by academic investigators who are not given protected time for their clinical investigator responsibilities and therefore lack the availability to meet with study clinical research associates (CRAs) or complete essential trial-related administrative duties. AMCs and ARSs can strengthen their participation and competitiveness in clinical research by:
o Allowing the use of central Institutional Review Boards (IRBs)
o Implementing a standardized clinical trial agreement format throughout the institution, ensuring its structure and key clauses reflect those commonly used by Clinical Research Organizations (CROs) and non-academic clinical research centers
o Setting reasonable budgets based on industry standards
o Allowing ARS/AMC medical investigators to have enough time to properly fulfill their clinical trial duties and responsibilities
By adopting these practices, institutions can reduce administrative costs and enhance their ability to attract and execute clinical trials.
Serious Adverse Event (SAE) reporting may be complicated. Whenever possible, investigators should report a clinical diagnosis rather than individual symptoms, as diagnoses provide clearer context for safety assessment and regulatory reporting.[3] If additional SAEs or AEs occur during a hospitalization, these should be reported as separate AEs (e.g., if a hospitalized participant develops acute pneumonia or needs a blood transfusion for anemia that developed while hospitalized).
Ensuring Clear and Consistent Safety Reporting
ARSs and AMCs should apply a conservative, ethics-driven approach to safety reporting. These institutions can preserve their gold-standard ethical and scientific safety culture while materially improving speed, consistency, and data quality by standardizing attribution, professionalizing safety roles, and enforcing internal timeliness metrics. Regulatory timelines must be met.

Staying Current with Evolving FDA Guidance
The FDA and other regulatory bodies are constantly modifying by issuing new rules and guidelines. It is important to keep updated on these changes. One free easy-to-use source for this is the FDA website: https://www.fda.gov/drugs/guidances-drugs/newly-added-guidance-documents
Medical Affairs
Strengthening Academic and Clinical Partnerships
Affiliating with academic institutions offers a highly effective way to engage with key opinion leaders (KOLs) across diverse disciplines. Such collaboration may occur through adjunct and clinical faculty appointments, or by volunteering to contribute expertise to didactic teaching or clinical training activities.
Here are some of the methods that medical affairs professionals can pursue to demonstrate expertise in a scientific area:
o Roles at scientific meetings
§ Poster presentations
§ Serving on conference panels
§ Podium presentations
§ Serving on a panel discussion
o Scientific journals
§ Publish editorials and manuscripts
§ Serve as manuscript reviewers
§ Serve on journal editorial boards
o Academic institutions
§ Voluntary staff appointments
§ Other affiliations such as with prominent nonprofit research facilities, FDA, or Veterans Association Hospitals
§ Formal relationships, e.g., adjunct, or clinical faculty
Advancing Shared Goals through Collaboration
Collaborations between medical affairs and academic programs are essential for driving shared progress across the industry
o Facilitating coordinated responses to regulatory changes that support broad industry improvement
o Publicizing important safety concerns to protect patients and stakeholders
o Advancing the integration of emerging technology
o Enhancing healthcare outcomes by promoting innovative, evidence-based solutions for society
Developing medical diagnostic and therapeutic guidelines
o Creating objective cooperative work forces
§ Also done with scientific organizations
o Helping to design best practices
Establishing non-profit associations to promote a specific disease or patient population
o Providing time and materials can produce win-win results for all
[1] WCG 2024 Clinical Research Site Challenges Report
[2] Lai J, et al. Drivers of Start-Up Delays in Global Randomized Clinical Trials. Ther Innov Regul Sci. 2021;55(1):212–223.
[3] FDA: Safety Reporting Requirements for Investigational New Drugs (INDs) and Bioavailability (BA)/Bioequivalence (BE) Studies. Guidance for Industry. 2012.
Safety and Efficacy of LP-10 Liposomal Tacrolimus in Oral Lichen Planus: A Multicenter Phase 2 Trial

Michael T Brennan 1, Jennifer Frustino 2, Kamal Al-Eryani 3, Herve Sroussi 4, Jennifer L Parish 5, Hirak B Routh 5, Sunil Dhawan 6, Gerald L Klein 7, Michael B Chancellor 8, Alessandro Villa 9
Affiliations
1 Department of Oral Medicine/Oral and Maxillofacial Surgery, Wake Forest University School of Medicine, Atrium Health Carolinas Medical Center, Charlotte, NC, USA.
2 Erie County Medical Center, Buffalo, NY, USA.
3 University of California San Francisco, San Francisco, CA, USA.
4 Brigham and Women's Hospital, Boston, MA, USA.
5 Paddington Testing Co, Inc, Philadelphia, PA, USA.
6 Center for Dermatology Clinical Research, Inc, Fremont, CA, USA.
7 MedSurgPI LLC, Brody School of Medicine, ECU, Franklinton, NC, USA.
8 Lipella Pharmaceuticals, Inc., Suite 505, 7800 Susquehanna, Pittsburgh, PA, 15208, USA. Michael.chancellor@lipella.com.
9 Oral Medicine and Oral Oncology Miami Cancer Institute, Baptist Health South Florida, Miami, FL, USA.
PMID: 41174333. DOI: 10.1007/s13555-025-01572-2
Abstract
Introduction: Oral lichen planus (OLP) is a serious chronic inflammatory condition with malignant transformation potential affecting six million Americans which has no US Food and Drug Administration (FDA)-approved therapy. LP-10, a novel liposomal tacrolimus oral rinse, overcomes the poor adherence to oral surfaces and inconsistent drug delivery limitations of existing treatments.
Methods: This phase 2a multicenter dose-ranging study evaluated LP-10 safety and efficacy in symptomatic OLP. Twenty-seven adults (22 female, 5 male) received a 10-mL oral rinse of LP-10 at 0.25 mg, 0.5 mg, or 1.0 mg of tacrolimus, for 3 min twice daily for 4 weeks. Safety assessments included monitoring of adverse events, laboratory studies, and tacrolimus blood levels. Efficacy was measured by investigator global assessment (IGA), reticulation/erythema/ulceration (REU) score, pain/sensitivity numerical rating scale (NRS), OLP symptom severity measure (OLPSSM), and patient global response assessment (GRA).
Results: All participants completed treatment without discontinuation or serious adverse events. Treatment-related treatment-emergent adverse events were mild/moderate (50 mild, 4 moderate). LP-10 demonstrated exceptional pharmacokinetic safety with mean tacrolimus levels remaining < 1.0 ng/mL in 75% of post-baseline measurements; the maximum individual level (4.5 ng/mL) remained well below the toxicity threshold (15 ng/mL) suggesting little absorbance into the blood. All efficacy endpoints showed statistically significant and clinically meaningful improvements at week 4: mean and standard deviation (SD) values for IGA decreased from 3.5 ± 0.51 to 1.8 ± 1.37 (p < 0.0001), pain NRS from 6.8 ± 1.90 to 2.3 ± 2.53 (p < 0.0001), sensitivity NRS from 7.2 ± 1.71 to 2.9 ± 2.29, REU from 26.5 ± 10.4 to 13.2 ± 8.15 (p < 0.0001). Of the 23 participants who responded to the GRA, approximately 78% participants reported that their quality of life was "moderately better" or "very much better" as compared to when they started the study, across all doses. All prior corticosteroid failures (5/5) responded, with benefits sustained through 2-week follow-up.
Conclusions: LP-10 demonstrated excellent safety with minimal systemic absorption and clinically meaningful efficacy, representing substantial improvement over existing therapies for this serious condition with significant unmet medical need. Larger controlled studies are warranted to confirm these promising findings.
Clinical trial registration: NCT06233591.
Keywords: Dose-ranging study; Liposome; Oral lichen planus; Oral mucosa; Phase 2 clinical trial; Tacrolimus.
Practical Pointers for Drug Development and Medical Affairs
Authors:
MedSurgPI: Gerald L. Klein, MD; Roger E. Morgan, MD; Freddy Byrth; Marion Stamp-Cole, Stephen Haworth, MD
SMART BioWorks: Eric Hacherl, PhD
Exquisite Biomedical Consulting: Shabnam Vaezzadeh, MD
Global Life Sciences Alliance: Denise McNerney
Independent DSMB Coordinator: Jamie Lazar
LBF BioPharma Consulting: Larry Florin
The Brown Group: Gail Brown, MD

Drug Development
New Guidelines from ICH E6(R3)
The International Council for Harmonisation (ICH) Guideline E6(R3), Good Clinical Practice revision represents a pivotal step toward modernizing clinical trial conduct and oversight. This update redefines how sponsors, investigators, and Medical Affairs teams manage quality and risk, emphasizing participant protection, data integrity, and responsible use of digital technologies.
1. Dynamic Risk Assessments (DRA): From Static to Adaptive Oversight
ICH E6(R3) suggests that sponsors develop a risk-based approach to clinical trial/development oversight, with the intensity and focus of activities such as monitoring, data review, and auditing adjusted based on the clinical trial’s complexity and potential impact on the study participants. Importantly, R3 builds upon prior versions of ICH E6 that emphasized Quality by Design (QbD), reinforcing that proactive, design-level quality principles should guide how risks are identified, prioritized, and managed throughout the study lifecycle.
Continuous evolution: These risk assessments should be periodically reviewed and, if needed, updated/changed based on the availability of new/additional data, changes in sites (number, geographic location, operational instructions, etc.), or technology (systems, site-facing interfaces, etc.) that may introduce new risks.
Adaptive monitoring: Replace standardized monitoring plans with real-time, adaptive strategies triggered by defined indicators:
Protocol deviations
Missing or inconsistent data
Emerging safety trends or unexpected adverse events
Independent oversight: Medical monitors and Data Safety Monitoring Boards (DSMBs) play a greater role in interpreting risk signals and ensuring participant safety, extending beyond passive review to include adaptive, data-informed interventions.
This shift aligns with regulatory trends emphasizing quality by design (QbD), risk-based monitoring (RBM), and fit-for-purpose quality systems—concepts central to ICH E8(R1) and E6(R3) alignment. At the heart of this evolution is a focus on “Critical-to-Quality” (CtQ) factors, as oversight activities should concentrate on the processes and data that are essential to assuring participant safety and the accumulation of reliable, interpretable patient data. A key crux of E6(R3) is the shift away from traditional, exhaustive source data verification (SDV) toward data-driven, centralized (statistical) monitoring strategies with a keen focus on critical data.
2. Reaffirming Participant Safety, Rights, and Well-Being
ICH E6(R3) reaffirms its ethical foundation: participant protection is paramount. Beyond compliance, sponsors must ensure participants are fully informed, empowered, and respected throughout the trial.
Transparency and accessibility: Investigators must have real-time access to new safety data, protocol amendments, and risk communications to ensure timely action at the site level.
Participant communication: Sponsors and Clinical Research Organizations (CROs) must provide safety information that is clear, understandable, and culturally appropriate, supporting autonomy and continued informed consent.
Ethical obligation: Sponsors are responsible not only for data collection but for safeguarding trust which is crucial for sustaining public confidence in research.
3. Digital Tools: From Operational Efficiency to Ethical Rigor
The guideline acknowledges the growing role of digital and decentralized technologies but reframes their purpose beyond efficiency and cost containment to ensure scientific validity and ethical implementation.
Fit-for-purpose validation: All digital tools (ePRO, wearables, AI analytics) must undergo formal validation to confirm they accurately capture, store, and transmit data as intended.
Ethical use and governance: Establish clear frameworks for data privacy, participant consent, and system reliability, particularly when technology intermediates participant interaction.
Integration with quality management: Digital systems should integrate seamlessly into the trial's risk-based quality framework to reinforce, not replace, scientific and ethical rigor.
Medical Affairs
Upholding Oversight and Quality
1. Medical Affairs (MA) teams serve as the interface between science, ethics, and communication, ensuring post-marketing and real-world activities align with ICH E6(R3) expectations. As AI becomes embedded in literature surveillance, scientific communication, medical review, and real-world data workflows, MA teams must ensure these tools are used responsibly, transparently, and in alignment with ICH E6(R3)’s quality and data-integrity principles.
2. System validation and data integrity: All computerized systems under MA control must be fully validated with robust audit trails, user-access controls, and metadata capture (author, reviewer, approver, time/date stamps). This applies to:
Key Opinion Leader (KOL) and stakeholder tracking (Customer Relationship Management) platforms
Publication planning and review systems
Real-world evidence and registry databases
AI-assisted content generation, literature monitoring, and medical insight analytics tools
3. Vendor oversight and accountability: Contracts with scientific vendors, communications agencies, and publication partners should clearly define:
Scope of work and deliverables
Delegated responsibilities and accountability
Oversight structure (Medical Affairs vs. vendor roles)
Performance metrics: timeliness, data accuracy, conflict-of-interest management, GxP compliance
Vendor training requirements: GCP, data privacy, and quality expectations
Clearly define Intellectual Property (IP) and practices to maintain IP protections
4. Documentation and training: Maintain oversight logs documenting all delegated activities and ensure team members receive training in essential areas, including, but not limited to:
Vendor selection and qualification criteria
Delegation and accountability management
Vendor audits and quality oversight procedures ensuring that participant care and safety decisions are made by competent professionals
Scope of work, deliverables, and agreed Key Performance Indicators (KPI)s
These practices demonstrate due diligence and establish clear traceability—key principles reinforced in ICH E6(R3).
Key Takeaway
ICH E6(R3) is not merely a compliance exercise; it's a call to rethink trial oversight as a living system. The guideline promotes balance between flexibility and accountability, urging organizations to align quality, ethics, and innovation at every stage of the trial lifecycle. For sponsors, CROs, and Medical Affairs teams, success depends on transparency, adaptability, and a proactive approach to risk management—hallmarks of modern, patient-centered research.
Practical Pointers for Drug Development / September 2025

Creating SOPs for Clinical Research Organizations (CROs), Site Networks, and Individual Clinical Research Sites
Standard Operating Procedures (SOPs) are foundational documents in drug development. They demonstrate regulatory compliance and provide practical, usable guidance for daily operations. Sponsors often request SOP master lists when evaluating vendors, and regulatory agencies regularly inspect them for adherence. A well-constructed SOP framework ensures consistency, clarity, and quality across functions, ultimately safeguarding inspection-readiness across CROs, site networks and investigator sites.
Key Principles:
· Regulatory and compliance alignment: SOPs must comply with the International Council for Harmonisation) ICH Good Clinical Practice (GCP) E6[R3], U.S. FDA regulations (21 CFR 11, 50, 54, 56, 312, 812, 314, 320), and regional requirements (e.g., European Medicines Agency (EMA), Medicines and Healthcare products Regulatory Agency (MHRA), and Pharmaceuticals and Medical Devices Agency (PMDA)). Device-related research should also align with the International Organization for Standardization (ISO) 14155:2020 and European Union Medical Device Regulation (EU MDR) 1027/745.
· Usability: SOPs must be practical, written in plain language with stepwise instructions, and include clear responsibilities tailored to job roles. Use process flowcharts, stepwise instructions, and checklists for complex workflows.
· Risk-based approach: In line with ICH E6(R3) and TransCelerate Risk-Based Quality Management (RBQM) frameworks, SOPs should integrate proactive risk identification, escalation pathways, and corrective and preventive action (CAPA) mechanisms to prevent deviations and strengthen quality-by-design.
· Organizations should regularly review overall business performance in conjunction with data on SOP adherence, deviations, and quality issues. Particular attention should be paid to occurrences of quality issues that are noted in combination with SOP adherence.
Structured Outline:
The SOP master list should include:
SOP title and number
Objective and owner
Version status and next review date
Clear index and categorization by function
Standardized SOP format:
Header/control information (ID, version, effective date, approval signatures)
Purpose and scope
Roles/responsibilities
Definitions, abbreviations, and acronyms
Stepwise procedures
Documentation/records (forms, logs, systems)
References
Version control
Practicality and usability
Flowcharts for complex processes
Checklists and templates for consistency
Sufficient detail and clarity without unnecessary complexity
Training modules integrated with SOP acknowledgements linked to specific job roles
Alignment of quality management systems (QMS)
References to SOPs for supporting processes/activities
References to process-specific training, which is indicated as necessary
Audit and continuous improvement
Routine internal audits to test compliance
Update processes when they are no longer practical
Ensure logical coverage across functions (safety reporting, data management, vendor oversight)
Regulatory Update - ICH E6(R3)
The recently finalized ICH E6(R3) Good Clinical Practice guidance emphasizes:
Greater flexibility for modern trial designs and diverse data sources
Integration of quality-by-design and risk-based quality management embedded in trial conduct
Clarification of sponsor and investigator responsibilties
Promotion of proportionality, relevance, and critical thinking across the trial lifecycle.
These principles should inform and shape all SOPs going forward, ensuring documents are not just compliant but also fit-for-purpose, risk-aware, digitally integrated, and globally harmonized.
Medical Affairs
Where to Publish Your Manuscript
Selecting the right journal for manuscript submission is a process that continues to grow in complexity with competing priorities of compliance, scientific impact, accessibility, speed and cost. Authors should begin by defining their intended audience and clarifying their main objective: maximizing impact, scientific prestige, influencing clinical practice or ensuring broad reach.
Key Considerations:
Audience: who are you trying to reach: researchers, clinicians, regulators or broader stakeholders?
Indexing: is the journal indexed in PubMed, Google Scholar, Embase, Scopus, Web of Science or regional databases relevant to your field?
Access model: open access vs. subscription
Publication fee: article processing charges, waivers or sponsor support
Journal Quality and Integrity:
Reputation and standing in the field
Metrics: impact factor, h-index, citation half-life, and Altmetric scores (used responsibly)
Peer-review rigor
Fit with the journal’s aims and scope
Familiarity with editorial board: consider reaching out to the editor prior to submission to gauge their interest in publishing your manuscript in their journal. Detail why this paper would be of interest to their readers.
Does it align with the scientific platform and publication plan?
Does it offer congress tie-ins?
Is it a special issue or themed collection in the area of expertise?
Beware of fraudulent journals reaching out under disingenuous purposes (e.g. click bait, obtaining a fee when a journal does not exist).
Consider a membership in Committee or Publication Ethics (COPE) or Director of Open Access Journals (DOAJ) to avoid a predatory journal.
Practical Issues:
Acceptance rate
Time to first decisions/time to publication
Availability of special issues, fast track, or online-ahead-of-print options
Ethics and Compliance:
Alignment with Good Publication Practice (GPP2022) and the International Committee of Medical Journal Editors (ICMJE) authorship criteria
Requirements for conflict-of-interest disclosures, trial registration (e.g., ClinicalTrials.gov) and data-sharing policies
In Summary: The choice of journal should align with the scientific content, target audience and strategic communications objectives. Balancing these factors often determines the real-world reach and visibility of your work. High-impact journals may provide prestige but at the cost of lower acceptance rates and longer timelines, while more specialized or open-access journals may offer faster publication and broader accessibility. Ultimately, the choice should align with both the scientific content and strategic communication objectives, compliance standards, and stakeholder needs. Balancing these considerations ensures that your work achieves both credibility and reach.
Published in Cureus: The Essential Role of Medical Monitors in Clinical Trials

New in Cureus: we outline how independent Medical Monitors safeguard patient safety, ensure regulatory alignment, and deliver unbiased, real-time oversight from protocol design through signal detection. Drawing on practical duties, escalation pathways, and cautionary case examples (e.g., first-in-human and CAR-T), we argue that medical monitoring is a strategic necessity—not just a compliance checkbox.
Navigating FDA meetings

Authors: Gerald L. Klein, MD+; Melissa Palmer, MD++; Freddy Byrth+; Roger E. Morgan, MD+; Thomas Krol, PharmD+; Seema Kumbhat, MD+++; Eric Hacherl, PhD++++, Shabnam Vaezzadeh, MD+++++
MedSurgPI+ Liver Consulting++ Ganglion Health+++ Smart Bio Works++++ Exquisite Biomedical Consulting+++++
Navigating FDA meetings is central to keeping drug, biologic, and device programs on track. This quick-reference chart highlights each meeting type’s purpose, timelines, and formality levels to help development teams plan more effectively and avoid delays