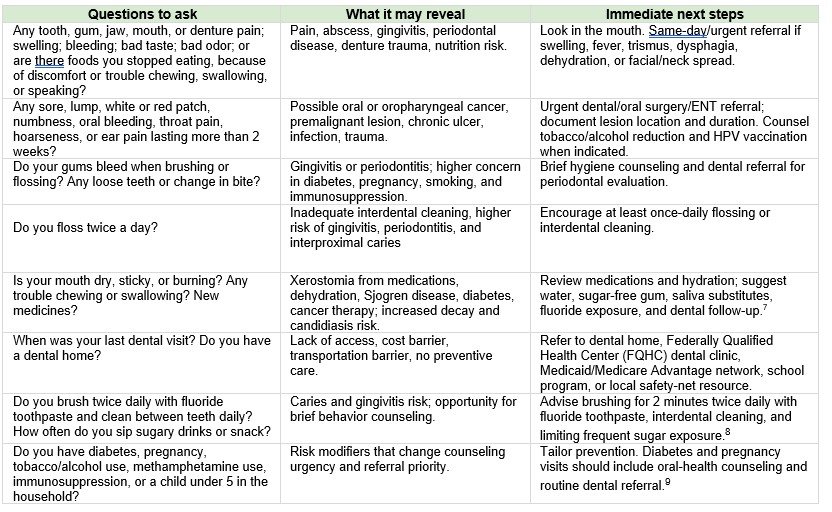
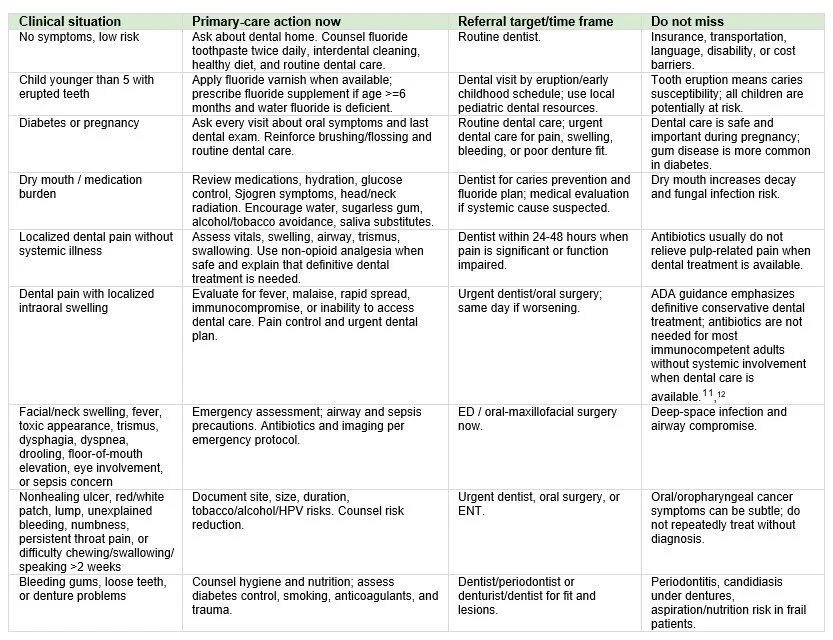
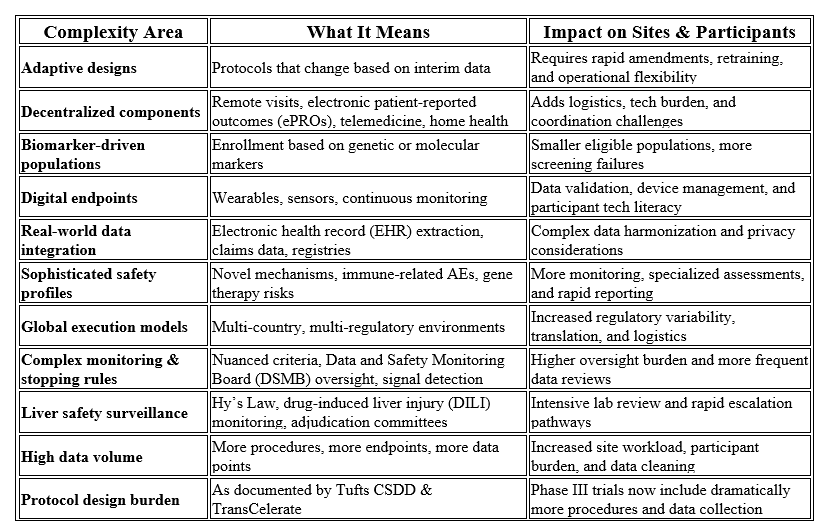
The East Carolina University (ECU) School of Dental Medicine is building its research enterprise.
This spotlight features a saliva Biobank at the ECU School of Dental Medicine as it builds its research enterprise under Dr. Alexandre R. Vieira, DDS, MS, PhD, Dean for Research
Authors: Gerald L. Klein, MD[1]; Freddy Byrth, BS[1]; Michael Fath, PhD[2]; Aida Carfagno, BS[3]; David Weinstein, MD, PhD[4]; Patrick Loebs, MSW, MSH[1]
Affiliation: MedSurgPI[1]; Cavabio Consulting[2]; Cedar and Stone Consulting[3]; David Weinstein Consulting[4]
Technology Innovation Spotlight features a saliva Biobank at the ECU School of Dental Medicine. Behind nearly every modern biomarker, companion diagnostic, and target-validation program sits an asset that seldom appears in the press release: well-characterized biospecimens, collected under controlled conditions and linked to longitudinal clinical data. Biobanking, the systematic collection, processing, storage, annotation, and distribution of biological specimens and their associated data, has quietly become one of the most consequential enabling technologies in drug, biologic, device, and diagnostic development. What was once thought of as “a freezer in the basement” is now a data-rich, standards-governed, increasingly automated infrastructure layer for precision medicine. Modern biobanks are therefore not only technical infrastructure; they are also trust infrastructure, dependent on transparent consent, privacy protections, and participant confidence that specimens and data will be used responsibly.
THE OPPORTUNITY
Saliva is among the most practical biofluids for population-scale collection. It can be gathered noninvasively, at low cost, and without specialized phlebotomy, and it carries a rich repertoire of analytes, including cells, nucleic acids, extracellular vesicles, metabolites, and proteins. Because oral and systemic health are increasingly understood to be connected, saliva offers a window not only into caries and periodontal disease but also into systemic conditions, with the added advantage of supporting repeated, longitudinal sampling at minimal patient burden.
THE INNOVATION
The center of gravity in biobanking has shifted from the specimen to the specimen plus the data wrapped around it. Three converging developments explain the change.
First, scale. Population-scale biobanks now operate at a size that was impractical a decade ago. The UK Biobank enrolled roughly 500,000 participants with linked genomic, imaging, and health-record data,[1] while the U.S. All of Us Research Program was designed explicitly to build a large, diverse cohort that mirrors the populations medicine actually serves.[2]
Second, data linkage. The research value of a biobank today depends heavily on how richly its specimens connect to electronic health records that can be queried in depth; robust phenotyping, and multi-omic readouts. The combination, not the specimen in isolation, is what enables agnostic and unbiased discovery across thousands of associations among diseases, genes, and exposures.[3]
Third, quality as a discipline. Preanalytical variables such as time to processing, freeze and thaw cycles, fixation, and storage temperature can materially alter the molecular characteristics of a sample and, with it, the validity of any downstream result. Reporting frameworks such as BRISQ (Biospecimen Reporting for Improved Study Quality) were created to make those variables transparent and reproducible,[4] and the international standard ISO 20387 now defines general requirements for biobanking competence and consistency.[5] Alongside scale and linkage, this quality science is what turns a collection of tubes into an extensive and rich research asset.

WHY IT MATTERS
• Noninvasive, low-cost collection enables large, diverse cohorts and repeat sampling that blood-based biobanks struggle to match.
• Chart-linked saliva supports biomarker discovery for oral conditions such as caries and periodontitis, as well as for systemic disease through the oral-systemic axis.
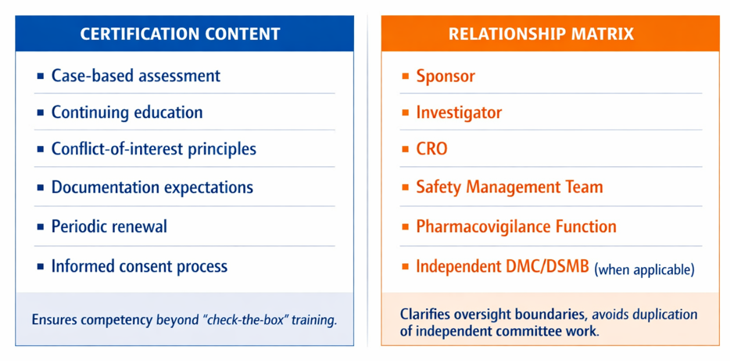
• A newer school building its portfolio can design consent scope, quality systems, and annotation for partnering from the outset rather than retrofitting them later.
• For sponsors, a well-governed saliva resource offers a practical platform for noninvasive biomarkers and companion-diagnostic work.
Representative large-scale biobanks frequently cited as exemplars:

FEATURED EXAMPLE:
Saliva is among the most practical biofluids for population-scale collection. It can be gathered noninvasively, at low cost, and without specialized phlebotomy, and it carries a rich repertoire of analytes, including cells, nucleic acids, extracellular vesicles, metabolites, and proteins. Because oral and systemic health are increasingly understood to be connected, saliva offers a window not only into caries and periodontal disease but also into systemic conditions, with the added advantage of supporting repeated, longitudinal sampling at minimal patient burden. ECU School of Dental Medicine is building its research enterprise under Dr. Alexandre R. Vieira, DDS, MS, PhD, Dean for Research, a dentist and human molecular geneticist whose work emphasizes characterizing patients’ whole health trajectories rather than studying one disease at a time. At his prior institution, Dr. Vieira established a dental-school registry under which patients entering the building were invited to contribute two things: permission to draw data from their records and a saliva sample. That pairing of biological specimen with chart-linked clinical data is the linkage that gives a modern biobank its translational power, and it is the model now being extended at ECU as the school formalizes a research portfolio supported by its Office of research facilities, including a Basic Science Laboratory, a Vivarium, and a Clinical Research Center.[1]
Why this matters to sponsors and licensees:
· Specimen quality is a data-quality problem: undocumented preanalytical variables undermine biomarker and companion-diagnostic claims.
· Consent scope is a gating factor: commercial and future-use permissions determine whether a collection can support a development program.
· ISO 20387 accreditation and BRISQ-style reporting are now table stakes in diligence, not nice-to-haves.
Diverse, electronic health record-linked cohorts shorten the path from hypothesis to validated, generalizable evidence.
THE COMMERCIAL AND TRANSLATIONAL OPPORTUNITY
Industry licensees and development partners do not value specimens generically. They value fit-for-purpose specimens. Four attributes drive whether a collection is commercially useful: documented provenance and preanalytical history; the depth and accuracy of clinical annotation; the scope of consent (in particular, whether commercial and future unspecified research use is permitted); and quality accreditation against an industry-recognized standard. Fit for purpose also depends on intended use: a collection that is adequate for exploratory biomarker discovery may not be sufficient for clinical validation, companion diagnostic development, or regulatory submission if chain of custody, preanalytical controls, assay validation, consent scope, or population representativeness are incomplete. A technically impressive collection with ambiguous consent or undocumented handling can be commercially unusable, an avoidable and expensive surprise late in a program.
For institutions that hold biobank assets, including universities, academic medical centers, and federal labs, this creates a real partnering opportunity, provided the asset is positioned in the language sponsors evaluate against. That framing is where many otherwise-strong collections under-realize their value.
REGULATORY, ETHICAL, AND QUALITY CONSIDERATIONS
Biobanking sits at the intersection of human-subjects protection, privacy law, and quality systems. In the United States, the revised Common Rule introduced provisions for broad consent covering storage, maintenance, and secondary research use of identifiable specimens and data,[1] while HIPAA governs the handling of protected health information. In the EU, GDPR imposes its own constraints on personal-data processing. Specimens used to support regulated product validation, particularly diagnostic and companion-diagnostic validation, additionally invite FDA scrutiny of provenance, characterization, and fitness for the intended use. Layered on top are quality and competence standards (ISO 20387) and BRISQ recognized biorepository best practices. The practical takeaway: governance and documentation decisions made at the moment of collection determine, years later, whether a specimen can be used at all.
MedSurgPI works at the intersection of clinical development, regulatory strategy, and Translational Medical Affairs, the same intersection where biobank value is won or lost. For institutions and inventors, we translate the science of a collection into the terms industry licensees actually evaluate: provenance, consent scope, fitness for purpose, and the regulatory path the specimens can support. For sponsors, we help define fit-for-purpose specimen and annotation requirements up front, scope consent to the development objective, and avoid the late-stage discovery that a collection cannot support the claim it was meant to enable. Medical Affairs adds particular value by defining the clinically meaningful questions a biobank should answer, identifying KOL and sponsor use cases, shaping evidence‑generation plans, and communicating both the potential and the limitations of the asset transparently.
Practical Pointer
A biobank’s translational ceiling is set at the moment of consent, collections built with commercial and future-use permissions from day one avoid the costly retrofitting that sidelines otherwise strong assets.

[1] Office for Human Research Protections, US Department of Health and Human Services. Federal Policy for the Protection of Human Subjects; Final Rule. Fed Regist. 2017;82(12):7149-7274. Codified at 45 CFR part 46.
[2] East Carolina University School of Dental Medicine. Research. Accessed June 9, 2026. https://dental.ecu.edu/research-department/
[3] Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. doi:10.1371/journal.pmed.1001779.
[4] All of Us Research Program Investigators. The “All of Us” Research Program. N Engl J Med. 2019;381(7):668-676. doi:10.1056/NEJMsr1809937.
[5] Beesley LJ, Salvatore M, Fritsche LG, Pandit A, Rao A, Brummett C, Willer CJ, Lisabeth LD, Mukherjee B. The emerging landscape of health research based on biobanks linked to electronic health records: existing resources, statistical challenges, and potential opportunities. Stat Med. 2020;39(6):773-800. doi:10.1002/sim.8445.
[6] Moore HM, Kelly AB, Jewell SD, McShane LM, Clark DP, Greenspan R, Hayes DF, Hainaut P, Kim P, Mansfield E, Potapova O, Riegman P, Rubinstein Y, Seijo E, Somiari S, Watson P, Weier HU, Zhu C, Vaught J. Biospecimen reporting for improved study quality (BRISQ). J Proteome Res. 2011;10(8):3429-3438. doi:10.1021/pr200021n.
[7] International Organization for Standardization. ISO 20387:2018. Biotechnology, Biobanking, General Requirements for Biobanking. Geneva, Switzerland: International Organization for Standardization; 2018.